EN
注册
登录
文献直达
发Paper
求职
问答
导师
期刊
资讯
首页
搜索
最新发布
我的收藏
清华大学/鄂尔多斯实验室魏飞-张晨曦团队在绿色航煤柔性合成领域取得重要进展
来源: X-MOL
2024-04-28
0
我要评论
取消
发布
上科大与碳谐科技联手JACS:拓展电合成氨领域
来源: X-MOL
2024-04-27
0
我要评论
取消
发布
“内外兼修”——铑掺杂实现铂合金超长氧还原电催化的调控策略
来源: X-MOL
2024-04-27
0
我要评论
取消
发布
黄振&何方Carbon Energy:原位透射电镜揭示NiFe2O4载氧体在化学链二氧化碳转化中晶格氧及金属离子的迁移扩散机理
来源: X-MOL
2024-04-26
0
我要评论
取消
发布
华东理工大学王灵芝/张金龙教授团队JACS:设计三相界面实现光催化甲烷选择性转化制乙醇
来源: X-MOL
2024-04-24
0
我要评论
取消
发布
单原子钌促进硫化镉光催化生物质衍生酸制备氨基酸
来源: X-MOL
2024-04-23
0
我要评论
取消
发布
化学所曹昌燕/中科大武晓君JACS:金属-硫界面,加氢反应新活性位点
来源: X-MOL
2024-04-21
0
我要评论
取消
发布
过渡态金属氧化物本征电催化析氧机制探究
来源: X-MOL
2024-04-21
0
我要评论
取消
发布
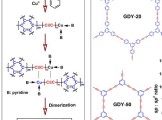
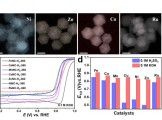
香港城市大学支春义教授团队Angew:调节石墨炔中的sp碳含量实现高效双氧水电合成
来源: X-MOL
2024-04-20
0
我要评论
取消
发布
厦门大学孙世刚课题组Angew:低温氢化合成高载量单原子Fe-N-C催化剂
来源: X-MOL
2024-04-19
0
我要评论
取消
发布
Angew:烧结诱导产生的晶界位点在提高铜基催化剂C-C偶联能力中的重要性
来源: X-MOL
2024-04-19
0
我要评论
取消
发布
Nano Res.[催化]│优化eg轨道电子实现钙钛矿氧化物电催化剂高效制氨
来源: X-MOL
2024-04-19
0
我要评论
取消
发布
清华大学章名田课题组 JACS Au|非对称双金属-氧化还原活性配体协同催化CO₂还原的机制研究
来源: JACS Au
2024-04-18
0
我要评论
取消
发布
Angew:原位谱学重新识别金属硫属催化剂在全水解反应中的活性物质及位点
来源: X-MOL
2024-04-18
0
我要评论
取消
发布
Nano Res.[催化]│青岛科技大学王磊教授团队: 铋纳米颗粒界面改性促进酸性CO₂电还原甲酸
来源: X-MOL
2024-04-18
0
我要评论
取消
发布
1
2
3
4
5
6
7
8
9
10
11
来看看大家都在关注些什么
有机化学
催化
材料
纳米科技
天然产物
高分子化学
药物与医疗
生命科学
无机化学
物理化学
理论和计算化学
分析化学
环境科学
食品与日用品
工业与商业
职场生涯
轻松生活
热点资讯
RSC主编推荐
盘点
广告
down
wechat
bug
bug




































 京公网安备 11010802027423号
京公网安备 11010802027423号