npj Computational Materials ( IF 9.4 ) Pub Date : 2019-12-10 , DOI: 10.1038/s41524-019-0259-z Changning Niu , You Rao , Wolfgang Windl , Maryam Ghazisaeidi
|
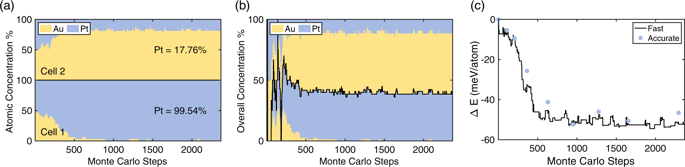
We propose a Multi-Cell Monte Carlo algorithm, or (MC)\({}^{2}\), for predicting stable phases in chemically complex crystalline systems. This algorithm takes advantage of multiple cells to represent possible phases, while eliminating the size and concentration restrictions in the previous counterparts. Free atomic transfer among cells is achieved via the application of the lever rule, where an assigned molar ratio virtually controls the percentage of each cell in the overall simulation, making (MC)\({}^{2}\) the first successful algorithm for simulating phase coexistence in crystalline solids. During the application of this method, all energies are directly computed via density functional theory calculations. We test the method by successful prediction of the stable phases of known binary systems. We then apply the method to a quaternary high-entropy alloy. The method is particularly powerful in predicting stable phases of multicomponent systems, for which phase diagrams do not exist.
中文翻译:
多单元蒙特卡洛方法进行相位预测
我们提出了一种多单元蒙特卡罗算法,或(MC)\({} ^ {2} \),用于预测化学复杂的晶体系统中的稳定相。该算法利用多个像元来表示可能的相,同时消除了先前对应物中的大小和浓度限制。单元格之间的自由原子转移是通过应用杠杆规则来实现的,其中杠杆比实际上控制了整个模拟中每个单元格的百分比,从而使(MC)\({} ^ {2} \)第一个成功的模拟结晶固体中相共存的算法。在该方法的应用过程中,所有能量都是通过密度泛函理论计算直接计算出来的。我们通过成功预测已知二进制系统的稳定相位来测试该方法。然后,我们将该方法应用于四元高熵合金。该方法在预测不存在相图的多组分系统的稳定相中特别有效。










































 京公网安备 11010802027423号
京公网安备 11010802027423号