Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
The mechanism of cancer drug addiction in ALK-positive T-Cell lymphoma.
Oncogene ( IF 6.9 ) Pub Date : 2019-12-05 , DOI: 10.1038/s41388-019-1136-4 Soumya S Rajan 1, 2 , Amit Dipak Amin 2, 3 , Lingxiao Li 2, 3 , Delphine C Rolland 4 , Haiquan Li 5 , Deukwoo Kwon 2, 6 , Mercedes F Kweh 2, 3 , Artavazd Arumov 1, 2 , Evan R Roberts 2 , Aimin Yan 2 , Venkatesha Basrur 7 , Kojo S J Elenitoba-Johnson 4 , Xi Steven Chen 2, 6 , Soham D Puvvada 8 , Yves A Lussier 8, 9 , Daniel Bilbao 2 , Megan S Lim 4 , Jonathan H Schatz 2, 3
Oncogene ( IF 6.9 ) Pub Date : 2019-12-05 , DOI: 10.1038/s41388-019-1136-4 Soumya S Rajan 1, 2 , Amit Dipak Amin 2, 3 , Lingxiao Li 2, 3 , Delphine C Rolland 4 , Haiquan Li 5 , Deukwoo Kwon 2, 6 , Mercedes F Kweh 2, 3 , Artavazd Arumov 1, 2 , Evan R Roberts 2 , Aimin Yan 2 , Venkatesha Basrur 7 , Kojo S J Elenitoba-Johnson 4 , Xi Steven Chen 2, 6 , Soham D Puvvada 8 , Yves A Lussier 8, 9 , Daniel Bilbao 2 , Megan S Lim 4 , Jonathan H Schatz 2, 3
Affiliation
|
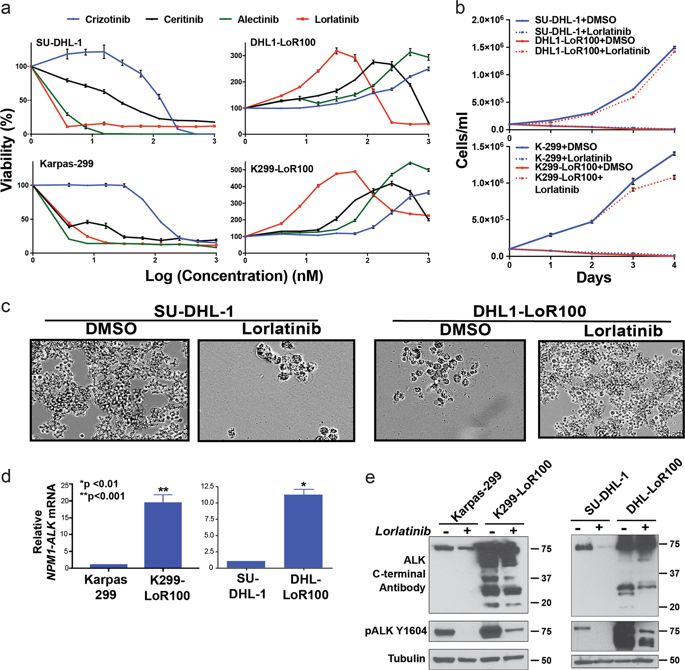
Rational new strategies are needed to treat tumors resistant to kinase inhibitors. Mechanistic studies of resistance provide fertile ground for development of new approaches. Cancer drug addiction is a paradoxical resistance phenomenon, well-described in MEK-ERK-driven solid tumors, in which drug-target overexpression promotes resistance but a toxic overdose of signaling if the inhibitor is withdrawn. This can permit prolonged control of tumors through intermittent dosing. We and others showed previously that cancer drug addiction arises also in the hematologic malignancy ALK-positive anaplastic large-cell lymphoma (ALCL) resistant to ALK-specific tyrosine kinase inhibitors (TKIs). This is driven by the overexpression of the fusion kinase NPM1-ALK, but the mechanism by which ALK overactivity drives toxicity upon TKI withdrawal remained obscure. Here we reveal the mechanism of ALK-TKI addiction in ALCL. We interrogated the well-described mechanism of MEK/ERK pathway inhibitor addiction in solid tumors and found it does not apply to ALCL. Instead, phosphoproteomics and confirmatory functional studies revealed that the STAT1 overactivation is the key mechanism of ALK-TKI addiction in ALCL. The withdrawal of TKI from addicted tumors in vitro and in vivo leads to overwhelming phospho-STAT1 activation, turning on its tumor-suppressive gene-expression program and turning off STAT3's oncogenic program. Moreover, a novel NPM1-ALK-positive ALCL PDX model showed a significant survival benefit from intermittent compared with continuous TKI dosing. In sum, we reveal for the first time the mechanism of cancer drug addiction in ALK-positive ALCL and the benefit of scheduled intermittent dosing in high-risk patient-derived tumors in vivo.
中文翻译:

ALK阳性T细胞淋巴瘤中癌症药物成瘾的机制。
需要合理的新策略来治疗对激酶抑制剂耐药的肿瘤。抗性机制研究为开发新方法提供了沃土。癌症药物成瘾是一种矛盾的耐药现象,在 MEK-ERK 驱动的实体瘤中得到了很好的描述,其中药物靶点的过度表达会促进耐药性,但如果停用抑制剂,则会产生毒性过量的信号传导。这可以允许通过间歇给药延长对肿瘤的控制。我们和其他人先前表明,癌症药物成瘾也出现在对 ALK 特异性酪氨酸激酶抑制剂 (TKI) 耐药的血液恶性肿瘤 ALK 阳性间变性大细胞淋巴瘤 (ALCL) 中。这是由融合激酶 NPM1-ALK 的过表达驱动的,但 ALK 过度活性在 TKI 退出时导致毒性的机制仍然不清楚。在这里,我们揭示了 ALCL 中 ALK-TKI 成瘾的机制。我们询问了 MEK/ERK 通路抑制剂成瘾在实体瘤中的充分描述机制,发现它不适用于 ALCL。相反,磷酸蛋白质组学和验证性功能研究表明,STAT1 过度激活是 ALCL 中 ALK-TKI 成瘾的关键机制。在体外和体内从成瘾性肿瘤中撤出 TKI 会导致大量的磷酸化 STAT1 激活,打开其肿瘤抑制基因表达程序并关闭 STAT3 的致癌程序。此外,与连续 TKI 给药相比,一种新型 NPM1-ALK 阳性 ALCL PDX 模型显示间歇性给药具有显着的生存益处。总共,
更新日期:2019-12-05
中文翻译:
ALK阳性T细胞淋巴瘤中癌症药物成瘾的机制。
需要合理的新策略来治疗对激酶抑制剂耐药的肿瘤。抗性机制研究为开发新方法提供了沃土。癌症药物成瘾是一种矛盾的耐药现象,在 MEK-ERK 驱动的实体瘤中得到了很好的描述,其中药物靶点的过度表达会促进耐药性,但如果停用抑制剂,则会产生毒性过量的信号传导。这可以允许通过间歇给药延长对肿瘤的控制。我们和其他人先前表明,癌症药物成瘾也出现在对 ALK 特异性酪氨酸激酶抑制剂 (TKI) 耐药的血液恶性肿瘤 ALK 阳性间变性大细胞淋巴瘤 (ALCL) 中。这是由融合激酶 NPM1-ALK 的过表达驱动的,但 ALK 过度活性在 TKI 退出时导致毒性的机制仍然不清楚。在这里,我们揭示了 ALCL 中 ALK-TKI 成瘾的机制。我们询问了 MEK/ERK 通路抑制剂成瘾在实体瘤中的充分描述机制,发现它不适用于 ALCL。相反,磷酸蛋白质组学和验证性功能研究表明,STAT1 过度激活是 ALCL 中 ALK-TKI 成瘾的关键机制。在体外和体内从成瘾性肿瘤中撤出 TKI 会导致大量的磷酸化 STAT1 激活,打开其肿瘤抑制基因表达程序并关闭 STAT3 的致癌程序。此外,与连续 TKI 给药相比,一种新型 NPM1-ALK 阳性 ALCL PDX 模型显示间歇性给药具有显着的生存益处。总共,










































 京公网安备 11010802027423号
京公网安备 11010802027423号