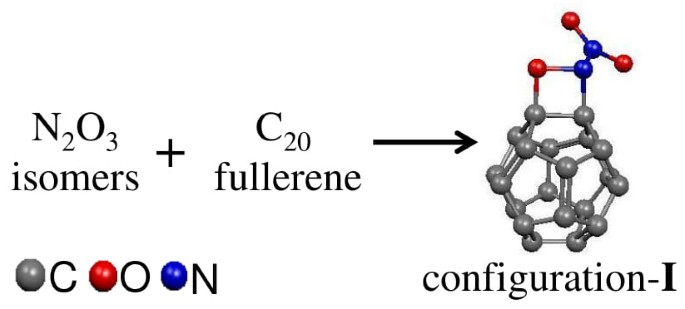
Structural and electronic properties of N2O3 adsorbed on C20 fullerene were investigated by Density Functional Theory. Before N2O3 was adsorbed on C20 fullerene surface, the structural properties of the isomers of the N2O3 were calculated. According to the total energy calculations, asym-N2O3 is the most stable isomer since it has lower energy than the other two isomers. Data obtained for the structural properties of N2O3 molecule are in agreement with the previous studies. After full structural optimization without any restrictions, the most stable structures were obtained. The adsorption energies with no dissociation of N2O3 were in the range of −2.88 to −3.55 eV for LDA and −2.02 to −2.45 eV for GGA. On the other hand, the dissociative adsorption yields a lower total energy with Eads values of −3.72 and −2.93 eV in LDA and GGA, respectively. According to these values, adsorption can be evaluated as chemisorption for all stable structures. The adsorption occurs with no reaction barriers except for one configuration. Although the co-adsorption of NO and NO2 molecules on the same fullerene is energetically less favorable compared to their adsorptions on separate fullerenes, the dissociative co-adsorption occurs with no energy barrier. The electronic structures are dominated by charge transfer from the fullerene to the adsorbate. The obtained HOMO-LUMO gap (GapHL) values are in the range of 1.02 and 1.35 eV for both LDA and GGA, respectively. The results presented here can be expected to guide future experimental and theoretical studies as new hybrid material.
Graphical abstract
中文翻译:
DFT研究C 20富勒烯吸附N 2 O 3的结构和电子性质
摘要
通过密度泛函理论研究了吸附在C 20富勒烯上的N 2 O 3的结构和电子性质。在将N 2 O 3吸附在C 20富勒烯表面上之前,计算出N 2 O 3的异构体的结构性质。根据总能量计算,asym -N 2 O 3是最稳定的异构体,因为它比其他两种异构体具有更低的能量。N 2 O 3的结构特性数据分子与先前的研究一致。经过全面的结构优化,没有任何限制,便获得了最稳定的结构。N 2 O 3没有解离的吸附能对于LDA为-2.88至-3.55 eV,对于GGA为-2.02至-2.45 eV。在另一方面,所述解离吸附产生具有较低的总能量ê广告中分别LDA和GGA,-3.72和-2.93电子伏特的值。根据这些值,可以将吸附评估为所有稳定结构的化学吸附。除了一种配置以外,没有反应障碍地发生吸附。虽然NO和NO 2共同吸附与它们在单独富勒烯上的吸附相比,在同一个富勒烯上的分子在能量上较不利,发生离解共吸附且没有能垒。电子结构由从富勒烯到被吸附物的电荷转移控制。对于LDA和GGA,获得的HOMO-LUMO间隙(GapHL)值分别在1.02和1.35 eV的范围内。此处提出的结果有望作为新的混合材料指导未来的实验和理论研究。












































 京公网安备 11010802027423号
京公网安备 11010802027423号