Molecular Diversity ( IF 3.9 ) Pub Date : 2020-08-29 , DOI: 10.1007/s11030-020-10136-9 Asma Khurshid 1 , Aamer Saeed 1 , Zaman Ashraf 2 , Qamar Abbas 3 , Mubashir Hassan 4
|
Abstract
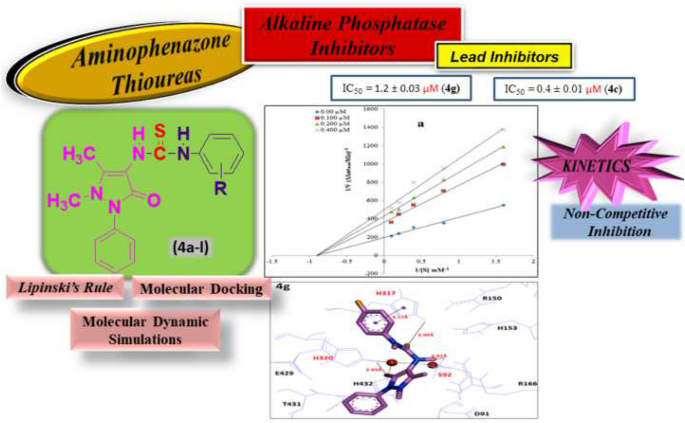
The work presented in this paper aims toward the synthesis of aryl thiourea derivatives 4a–l of pyrazole based nonsteroidal anti-inflammatory drug named 4-aminophenazone, as potential inhibitors of intestinal alkaline phosphatase enzyme. The screening of synthesized target compounds 4a–l for unraveling the anti-inflammatory potential against calf intestinal alkaline phosphatase gives rise to lead member 4c possessing IC50 value 0.420 ± 0.012 µM, many folds better than reference standard used (KH2PO4 IC50 = 2.8 ± 0.06 µM and l-phenylalanine IC50 = 100 ± 3.1 µM). SAR for unfolding the active site binding pocket interaction along with the mode of enzyme inhibition based on kinetic studies is carried out which showed non-competitive binding mode. The enzyme inhibition studies were further supplemented by molecular dynamic simulations for predicting the protein behavior against active inhibitors 4c and 4g during docking analysis. The preliminary toxicity of the synthesized compounds was determined by using brine shrimp assay. This work also includes detailed biochemical analysis along with RO5 parameters for all the newly synthesized drug derivatives 4a–l.
Graphic abstract
中文翻译:
通过辅助计算分子动力学模拟了解氨基吩酮衍生的芳基硫脲对肠碱性磷酸酶的酶抑制作用:合成、表征、SAR 和动力学分析。
摘要
本文中提出的工作旨在合成芳基硫脲衍生物4a – l的吡唑类非甾体抗炎药 4-氨基苯甲酮,作为肠道碱性磷酸酶的潜在抑制剂。筛选合成的目标化合物4a – l以揭示对小牛肠碱性磷酸酶的抗炎潜力,产生了具有 IC 50值 0.420 ± 0.012 µM 的先导成员4c,比使用的参考标准 (KH 2 PO 4 IC 50)好很多倍= 2.8 ± 0.06 µM 和l -苯丙氨酸 IC 50 = 100 ± 3.1 µM)。进行了基于动力学研究的用于展开活性位点结合口袋相互作用以及酶抑制模式的 SAR,其显示出非竞争性结合模式。酶抑制研究得到了分子动力学模拟的进一步补充,用于预测对接分析期间针对活性抑制剂4c和4g的蛋白质行为。合成化合物的初步毒性是通过盐水虾试验确定的。这项工作还包括详细的生化分析以及所有新合成的药物衍生物4a – l 的RO5 参数。










































 京公网安备 11010802027423号
京公网安备 11010802027423号