Departamento de Química e Física - Centro de Ciências Exatas, Naturais e da Saúde - CCENS, Universidade Federal do Espírito Santo, Alegre, Espírito Santo, 29500-000, Brazil.
Instituto de Física, Universidade Federal de Goiás, Av. Esperança, s/n - Campus Samambaia, Goiânia, GO, 74690-900, Brazil.
Bremen Center for Computational Materials Science, University of Bremen, P.O. Box 330440, 28334, Bremen, Germany.
Computational Science Research Center, Computational Science and Applied Research Institute Shenzhen, No.10 East Xibeiwang Road, 100193, Beijing, China.
Max Planck Institute for the Structure and Dynamics of Matter, Luruper Chaussee 149, Geb. 99 (CFEL), 22761, Hamburg, Germany.
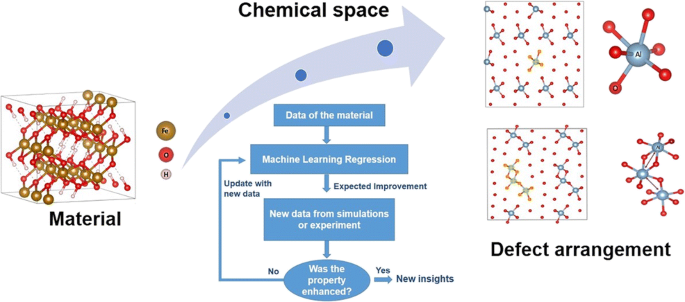
Designing and understanding the mechanism of non-stoichiometric materials with enhanced properties is challenging, both experimentally and even computationally, due to the large number of chemical spaces and their distributions through the material. In the current work, it is proposed a Machine Learning approach coupled with the Efficient Global Optimization (EGO) method—an Adaptive Design (AD)—to model local defects in materials from first-principle calculations. Our method takes into account the smallest sample set as possible, envisioning the material defect structure relationship with target properties for new insights. As an example, the AD framework allows us to study the stability and the structure of the modified goethite (Fe0.875Al0.125OOH) by considering a proper defect distribution, from first-principle calculations. The chemical space search for the modified goethite was evaluated by starting from different sizes and configurations of the samples as well as different surrogate models (ANN and Gaussian Process; GP), acquisition functions, and descriptors. Our results show that the same local solution of several defect arrangements in Fe0.875Al0.125OOH is found regardless of the initial sample and regression model. This indicates the efficiency of our search method. We also discuss the role of the descriptors in the accelerated global search for defects in material modeling. We conclude that the AD method applied in material defects is a successful approach in automating the search within huge chemical spaces from first-principle calculations by considering small samples. This method can be applied to mechanistic elucidation of non-stoichiometric materials, solid solutions, alloys, and Schottky and Frenkel defects, essential for material design and discovery.
Graphical abstract
中文翻译:
一种基于第一性原理计算的材料中缺陷分布建模的自适应设计方法。
由于大量的化学空间及其在整个材料中的分布,设计和理解具有增强特性的非化学计量材料的机理在实验甚至计算上都具有挑战性。在当前工作中,提出了一种将机器学习方法与有效全局优化(EGO)方法(一种自适应设计(AD))相结合的方法,以便根据第一性原理计算对材料中的局部缺陷进行建模。我们的方法考虑了最小的样本集,并设想了材料缺陷结构与目标属性之间的关系,以获取新的见解。例如,AD框架使我们能够研究改性针铁矿(Fe 0.875 Al 0.125通过第一原理计算,考虑适当的缺陷分布。通过从不同大小和构造的样品以及不同的替代模型(ANN和高斯过程; GP),采集函数和描述符开始,评估了改性针铁矿的化学空间搜索。我们的结果表明,Fe 0.875 Al 0.125中几种缺陷排列的相同局部解无论初始样本和回归模型如何,都可以找到OOH。这表明我们的搜索方法的效率。我们还将讨论描述符在加速全局搜索材料建模中的缺陷中的作用。我们得出的结论是,应用于材料缺陷的AD方法是一种成功的方法,可以通过考虑小样本,从第一性原理计算自动在巨大的化学空间内进行搜索。此方法可用于对非化学计量材料,固溶体,合金以及材料设计和发现必不可少的肖特基和弗伦克尔缺陷进行机械解析。
图形概要
 Graphical abstract
Graphical abstract
 Graphical abstract
Graphical abstract

























 京公网安备 11010802027423号
京公网安备 11010802027423号