Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Chromosomal assembly and analyses of genome-wide recombination rates in the forest pathogenic fungus Armillaria ostoyae
Heredity ( IF 3.1 ) Pub Date : 2020-03-13 , DOI: 10.1038/s41437-020-0306-z Renate Heinzelmann 1, 2 , Daniel Rigling 1 , György Sipos 3 , Martin Münsterkötter 3, 4 , Daniel Croll 5
Heredity ( IF 3.1 ) Pub Date : 2020-03-13 , DOI: 10.1038/s41437-020-0306-z Renate Heinzelmann 1, 2 , Daniel Rigling 1 , György Sipos 3 , Martin Münsterkötter 3, 4 , Daniel Croll 5
Affiliation
|
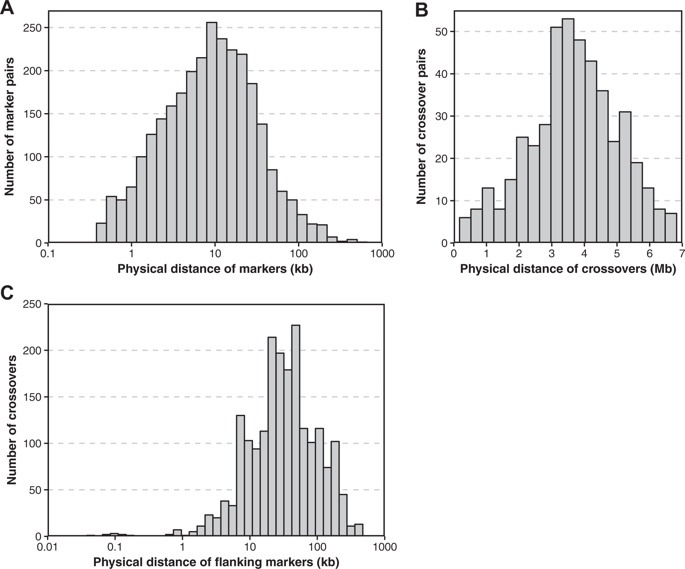
Recombination shapes the evolutionary trajectory of populations and plays an important role in the faithful transmission of chromosomes during meiosis. Levels of sexual reproduction and recombination are important properties of host–pathogen interactions because the speed of antagonistic co-evolution depends on the ability of hosts and pathogens to generate genetic variation. However, our understanding of the importance of recombination is limited because large taxonomic groups remain poorly investigated. Here, we analyze recombination rate variation in the basidiomycete fungus Armillaria ostoyae, which is an aggressive pathogen on a broad range of conifers and other trees. We analyzed a previously constructed, dense genetic map based on 198 single basidiospore progeny from a cross. Progeny were genotyped at a genome-wide set of single-nucleotide polymorphism (SNP) markers using double digest restriction site associated DNA sequencing. Based on a linkage map of on 11,700 SNPs spanning 1007.5 cM, we assembled genomic scaffolds into 11 putative chromosomes of a total genome size of 56.6 Mb. We identified 1984 crossover events among all progeny and found that recombination rates were highly variable along chromosomes. Recombination hotspots tended to be in regions close to the telomeres and were more gene-poor than the genomic background. Genes in proximity to recombination hotspots were encoding on average shorter proteins and were enriched for pectin degrading enzymes. Our analyses enable more powerful population and genome-scale studies of a major tree pathogen.
中文翻译:

森林病原菌蜜环菌的染色体组装和全基因组重组率分析
重组塑造了种群的进化轨迹,并在减数分裂过程中染色体的忠实传递中起着重要作用。有性繁殖和重组的水平是宿主-病原体相互作用的重要特性,因为对抗性共同进化的速度取决于宿主和病原体产生遗传变异的能力。然而,我们对重组重要性的理解是有限的,因为对大型分类群的研究仍然很少。在这里,我们分析了担子菌真菌 Armillaria ostoyae 的重组率变化,这是一种对多种针叶树和其他树木的侵袭性病原体。我们分析了先前构建的基于杂交的 198 个单个担子孢子后代的密集遗传图谱。使用双酶切限制位点相关的 DNA 测序,在全基因组单核苷酸多态性 (SNP) 标记集上对后代进行基因分型。基于跨越 1007.5 cM 的 11,700 个 SNP 的连锁图,我们将基因组支架组装成 11 条假定的染色体,总基因组大小为 56.6 Mb。我们在所有后代中确定了 1984 年的交叉事件,发现重组率沿染色体高度可变。重组热点往往位于靠近端粒的区域,并且比基因组背景更缺乏基因。靠近重组热点的基因平均编码较短的蛋白质,并富含果胶降解酶。我们的分析使对主要树木病原体进行更强大的种群和基因组规模研究成为可能。
更新日期:2020-03-13
中文翻译:
森林病原菌蜜环菌的染色体组装和全基因组重组率分析
重组塑造了种群的进化轨迹,并在减数分裂过程中染色体的忠实传递中起着重要作用。有性繁殖和重组的水平是宿主-病原体相互作用的重要特性,因为对抗性共同进化的速度取决于宿主和病原体产生遗传变异的能力。然而,我们对重组重要性的理解是有限的,因为对大型分类群的研究仍然很少。在这里,我们分析了担子菌真菌 Armillaria ostoyae 的重组率变化,这是一种对多种针叶树和其他树木的侵袭性病原体。我们分析了先前构建的基于杂交的 198 个单个担子孢子后代的密集遗传图谱。使用双酶切限制位点相关的 DNA 测序,在全基因组单核苷酸多态性 (SNP) 标记集上对后代进行基因分型。基于跨越 1007.5 cM 的 11,700 个 SNP 的连锁图,我们将基因组支架组装成 11 条假定的染色体,总基因组大小为 56.6 Mb。我们在所有后代中确定了 1984 年的交叉事件,发现重组率沿染色体高度可变。重组热点往往位于靠近端粒的区域,并且比基因组背景更缺乏基因。靠近重组热点的基因平均编码较短的蛋白质,并富含果胶降解酶。我们的分析使对主要树木病原体进行更强大的种群和基因组规模研究成为可能。










































 京公网安备 11010802027423号
京公网安备 11010802027423号