当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Isobaric Labeling Quantitative Metaproteomics for the Study of Gut Microbiome Response to Arsenic.
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2019-01-16 , DOI: 10.1021/acs.jproteome.8b00666 Chih-Wei Liu 1 , Liang Chi 1 , Pengcheng Tu 1 , Jingchuan Xue 1 , Hongyu Ru 2 , Kun Lu 1
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2019-01-16 , DOI: 10.1021/acs.jproteome.8b00666 Chih-Wei Liu 1 , Liang Chi 1 , Pengcheng Tu 1 , Jingchuan Xue 1 , Hongyu Ru 2 , Kun Lu 1
Affiliation
|
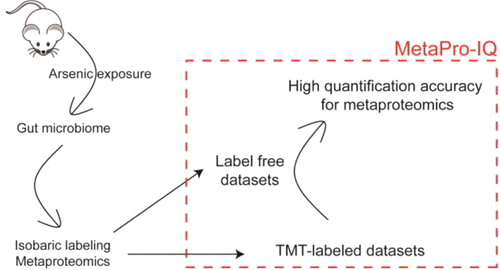
Quantitative metaproteomics is a relatively new research field that applies proteomics techniques to study microbial proteins of the microbiome and holds great potential in truly quantifying the functional proteins actually expressed by microbes in the biological environment, such as the gastrointestinal tract. The significant association between arsenic exposure and gut microbiome perturbations has been reported; however, metaproteomics has not yet been applied to study arsenic-induced proteome changes of the microbiome. Most importantly, to our knowledge, isobaric-labeling-based large-scale metaproteomics has not been reported using the advanced database-search approaches such as MetaPro-IQ and matched metagenome database-search strategies to provide high quantification accuracy and fewer missing quantification values. In the present study, a new experimental workflow coupled to isobaric labeling and MetaPro-IQ was demonstrated for the metaproteomics study of arsenic-induced gut microbiome perturbations. The advantages of this workflow were also discussed. For all 18 fecal samples analyzed, 7611 protein groups were quantified without any missing values. The consistent results of expression profiles were observed between 16S rRNA gene sequencing and metaproteomics. This isobaric-labeling-based workflow demonstrated the significant improvement of quantitative metaproteomics for gut microbiome study.
中文翻译:

等压标记定量元蛋白质组学,用于研究肠道微生物组对砷的反应。
定量元蛋白质组学是一个相对较新的研究领域,它应用蛋白质组学技术研究微生物组的微生物蛋白,在真正量化微生物在胃肠道等生物环境中实际表达的功能蛋白方面具有巨大的潜力。据报道,砷暴露与肠道微生物组扰动之间存在显着联系。然而,元蛋白质组学尚未用于研究砷诱导的微生物组蛋白质组变化。最重要的是,据我们所知,尚未使用高级数据库搜索方法(例如MetaPro-IQ和匹配的元基因组数据库搜索策略)提供基于等压标记的大规模元蛋白质组学,以提供较高的定量准确度和较少的缺失定量值。在目前的研究中,结合等压线标记和MetaPro-IQ的新实验工作流程已被证明用于砷诱导的肠道微生物组扰动的元蛋白质组学研究。还讨论了此工作流程的优点。对于分析的所有18个粪便样品,对7611个蛋白质组进行了定量分析,没有任何缺失值。在16S rRNA基因测序和元蛋白质组学之间观察到表达谱的一致结果。这种基于等压线标记的工作流程证明了肠道微生物组研究定量元蛋白质组学的显着改进。量化了7611个蛋白质组,没有任何遗漏值。在16S rRNA基因测序和元蛋白质组学之间观察到表达谱的一致结果。这种基于等压线标记的工作流程证明了肠道微生物组研究定量元蛋白质组学的显着改进。量化了7611个蛋白质组,没有任何遗漏值。在16S rRNA基因测序和元蛋白质组学之间观察到表达谱的一致结果。这种基于等压线标记的工作流程证明了肠道微生物组研究定量元蛋白质组学的显着改进。
更新日期:2019-02-07
中文翻译:
等压标记定量元蛋白质组学,用于研究肠道微生物组对砷的反应。
定量元蛋白质组学是一个相对较新的研究领域,它应用蛋白质组学技术研究微生物组的微生物蛋白,在真正量化微生物在胃肠道等生物环境中实际表达的功能蛋白方面具有巨大的潜力。据报道,砷暴露与肠道微生物组扰动之间存在显着联系。然而,元蛋白质组学尚未用于研究砷诱导的微生物组蛋白质组变化。最重要的是,据我们所知,尚未使用高级数据库搜索方法(例如MetaPro-IQ和匹配的元基因组数据库搜索策略)提供基于等压标记的大规模元蛋白质组学,以提供较高的定量准确度和较少的缺失定量值。在目前的研究中,结合等压线标记和MetaPro-IQ的新实验工作流程已被证明用于砷诱导的肠道微生物组扰动的元蛋白质组学研究。还讨论了此工作流程的优点。对于分析的所有18个粪便样品,对7611个蛋白质组进行了定量分析,没有任何缺失值。在16S rRNA基因测序和元蛋白质组学之间观察到表达谱的一致结果。这种基于等压线标记的工作流程证明了肠道微生物组研究定量元蛋白质组学的显着改进。量化了7611个蛋白质组,没有任何遗漏值。在16S rRNA基因测序和元蛋白质组学之间观察到表达谱的一致结果。这种基于等压线标记的工作流程证明了肠道微生物组研究定量元蛋白质组学的显着改进。量化了7611个蛋白质组,没有任何遗漏值。在16S rRNA基因测序和元蛋白质组学之间观察到表达谱的一致结果。这种基于等压线标记的工作流程证明了肠道微生物组研究定量元蛋白质组学的显着改进。










































 京公网安备 11010802027423号
京公网安备 11010802027423号