当前位置:
X-MOL 学术
›
Eur. J. Med. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Identification of DOT1L inhibitors by structure-based virtual screening adapted from a nucleoside-focused library.
European Journal of Medicinal Chemistry ( IF 6.0 ) Pub Date : 2020-01-02 , DOI: 10.1016/j.ejmech.2019.112023 Garrett S Gibbons 1 , Amarraj Chakraborty 2 , Sierrah M Grigsby 1 , Afoma C Umeano 3 , Chenzhong Liao 4 , Omar Moukha-Chafiq 5 , Vibha Pathak 5 , Bini Mathew 5 , Young-Tae Lee 4 , Yali Dou 4 , Stephan C Schürer 6 , Robert C Reynolds 7 , Timothy S Snowden 2 , Zaneta Nikolovska-Coleska 8
European Journal of Medicinal Chemistry ( IF 6.0 ) Pub Date : 2020-01-02 , DOI: 10.1016/j.ejmech.2019.112023 Garrett S Gibbons 1 , Amarraj Chakraborty 2 , Sierrah M Grigsby 1 , Afoma C Umeano 3 , Chenzhong Liao 4 , Omar Moukha-Chafiq 5 , Vibha Pathak 5 , Bini Mathew 5 , Young-Tae Lee 4 , Yali Dou 4 , Stephan C Schürer 6 , Robert C Reynolds 7 , Timothy S Snowden 2 , Zaneta Nikolovska-Coleska 8
Affiliation
|
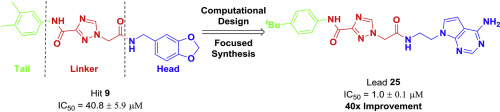
Disruptor of Telomeric Silencing 1-Like (DOT1L), the sole histone H3 lysine 79 (H3K79) methyltransferase, is required for leukemogenic transformation in a subset of leukemias bearing chromosomal translocations of the Mixed Lineage Leukemia (MLL) gene, as well as other cancers. Thus, DOT1L is an attractive therapeutic target and discovery of small molecule inhibitors remain of high interest. Herein, we are presenting screening results for a unique focused library of 1200 nucleoside analogs originally produced under the aegis of the NIH Pilot Scale Library Program. The complete nucleoside set was screened virtually against DOT1L, resulting in 210 putative hits. In vitro screening of the virtual hits resulted in validation of 11 compounds as DOT1L inhibitors clustered into two distinct chemical classes, adenosine-based inhibitors and a new chemotype that lacks adenosine. Based on the developed DOT1L ligand binding model, a structure-based design strategy was applied and a second-generation of non-nucleoside DOT1L inhibitors was developed. Newly synthesized compound 25 was the most potent DOT1L inhibitor in the new series with an IC50 of 1.0 μM, showing 40-fold improvement in comparison with hit 9 and exhibiting reasonable on target effects in a DOT1L dependent murine cell line. These compounds represent novel chemical probes with a unique non-nucleoside scaffold that bind and compete with the SAM binding site of DOT1L, thus providing foundation for further medicinal chemistry efforts to develop more potent compounds.
中文翻译:

DOT1L抑制剂的鉴定是通过对基于核苷的文库进行改编的基于结构的虚拟筛选来进行的。
端粒沉默1-Like(DOT1L)的破坏者是唯一的组蛋白H3赖氨酸79(H3K79)甲基转移酶,对于白血病的子集进行白血病生成转化时,需要进行混合谱系白血病(MLL)基因的染色体易位以及其他癌症。因此,DOT1L是有吸引力的治疗靶标,小分子抑制剂的发现仍然引起人们的极大兴趣。在此,我们展示了最初由NIH Pilot Scale Library Program主持下生产的1200个核苷类似物的独特聚焦文库的筛选结果。实际上针对DOT1L筛选了完整的核苷组,导致210个推定命中。由于DOT1L抑制剂聚集在两个不同的化学类别中,因此虚拟筛选的体外筛选结果验证了11种化合物的有效性,基于腺苷的抑制剂和缺乏腺苷的新化学型。基于已开发的DOT1L配体结合模型,应用了基于结构的设计策略,并开发了第二代非核苷DOT1L抑制剂。新合成的化合物25是新系列中最有效的DOT1L抑制剂,IC50为1.0μM,与命中9相比显示40倍的改善,并且在依赖DOT1L的鼠细胞系中表现出合理的靶作用。这些化合物代表了具有独特的非核苷支架的新型化学探针,该支架与DOT1L的SAM结合位点结合并竞争,从而为进一步开发更有效的化合物的药物化学研究奠定了基础。应用了基于结构的设计策略,并开发了第二代非核苷DOT1L抑制剂。新合成的化合物25是新系列中最有效的DOT1L抑制剂,IC50为1.0μM,与命中9相比显示40倍的改善,并且在依赖DOT1L的鼠细胞系中表现出合理的靶作用。这些化合物代表了具有独特的非核苷支架的新型化学探针,该支架与DOT1L的SAM结合位点结合并竞争,从而为进一步开发更有效的化合物的药物化学研究奠定了基础。应用了基于结构的设计策略,并开发了第二代非核苷DOT1L抑制剂。新合成的化合物25是新系列中最有效的DOT1L抑制剂,IC50为1.0μM,与命中9相比显示40倍的改善,并且在DOT1L依赖的鼠类细胞系中表现出合理的靶作用。这些化合物代表了具有独特的非核苷支架的新型化学探针,该支架与DOT1L的SAM结合位点结合并竞争,从而为进一步开发更有效的化合物的药物化学研究奠定了基础。与DOT1L依赖的鼠细胞系相比,与第9击相比显示出40倍的改善,并表现出合理的靶效应。这些化合物代表了具有独特的非核苷支架的新型化学探针,该支架与DOT1L的SAM结合位点结合并竞争,从而为进一步开发更有效的化合物的药物化学研究奠定了基础。与DOT1L依赖的鼠细胞系相比,与第9击相比显示出40倍的改善,并表现出合理的靶效应。这些化合物代表了具有独特的非核苷支架的新型化学探针,该支架与DOT1L的SAM结合位点结合并竞争,从而为进一步开发更有效的化合物的药物化学研究奠定了基础。
更新日期:2020-01-02
中文翻译:
DOT1L抑制剂的鉴定是通过对基于核苷的文库进行改编的基于结构的虚拟筛选来进行的。
端粒沉默1-Like(DOT1L)的破坏者是唯一的组蛋白H3赖氨酸79(H3K79)甲基转移酶,对于白血病的子集进行白血病生成转化时,需要进行混合谱系白血病(MLL)基因的染色体易位以及其他癌症。因此,DOT1L是有吸引力的治疗靶标,小分子抑制剂的发现仍然引起人们的极大兴趣。在此,我们展示了最初由NIH Pilot Scale Library Program主持下生产的1200个核苷类似物的独特聚焦文库的筛选结果。实际上针对DOT1L筛选了完整的核苷组,导致210个推定命中。由于DOT1L抑制剂聚集在两个不同的化学类别中,因此虚拟筛选的体外筛选结果验证了11种化合物的有效性,基于腺苷的抑制剂和缺乏腺苷的新化学型。基于已开发的DOT1L配体结合模型,应用了基于结构的设计策略,并开发了第二代非核苷DOT1L抑制剂。新合成的化合物25是新系列中最有效的DOT1L抑制剂,IC50为1.0μM,与命中9相比显示40倍的改善,并且在依赖DOT1L的鼠细胞系中表现出合理的靶作用。这些化合物代表了具有独特的非核苷支架的新型化学探针,该支架与DOT1L的SAM结合位点结合并竞争,从而为进一步开发更有效的化合物的药物化学研究奠定了基础。应用了基于结构的设计策略,并开发了第二代非核苷DOT1L抑制剂。新合成的化合物25是新系列中最有效的DOT1L抑制剂,IC50为1.0μM,与命中9相比显示40倍的改善,并且在依赖DOT1L的鼠细胞系中表现出合理的靶作用。这些化合物代表了具有独特的非核苷支架的新型化学探针,该支架与DOT1L的SAM结合位点结合并竞争,从而为进一步开发更有效的化合物的药物化学研究奠定了基础。应用了基于结构的设计策略,并开发了第二代非核苷DOT1L抑制剂。新合成的化合物25是新系列中最有效的DOT1L抑制剂,IC50为1.0μM,与命中9相比显示40倍的改善,并且在DOT1L依赖的鼠类细胞系中表现出合理的靶作用。这些化合物代表了具有独特的非核苷支架的新型化学探针,该支架与DOT1L的SAM结合位点结合并竞争,从而为进一步开发更有效的化合物的药物化学研究奠定了基础。与DOT1L依赖的鼠细胞系相比,与第9击相比显示出40倍的改善,并表现出合理的靶效应。这些化合物代表了具有独特的非核苷支架的新型化学探针,该支架与DOT1L的SAM结合位点结合并竞争,从而为进一步开发更有效的化合物的药物化学研究奠定了基础。与DOT1L依赖的鼠细胞系相比,与第9击相比显示出40倍的改善,并表现出合理的靶效应。这些化合物代表了具有独特的非核苷支架的新型化学探针,该支架与DOT1L的SAM结合位点结合并竞争,从而为进一步开发更有效的化合物的药物化学研究奠定了基础。










































 京公网安备 11010802027423号
京公网安备 11010802027423号