当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
A first principles study of the spin-orbit coupling effect in LiM (M = Na, K, Rb, Cs) molecules.
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2020-01-13 , DOI: 10.1039/c9cp06421d S V Kozlov 1 , E A Bormotova 1 , A A Medvedev 1 , E A Pazyuk 1 , A V Stolyarov 1 , A Zaitsevskii 2
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2020-01-13 , DOI: 10.1039/c9cp06421d S V Kozlov 1 , E A Bormotova 1 , A A Medvedev 1 , E A Pazyuk 1 , A V Stolyarov 1 , A Zaitsevskii 2
Affiliation
|
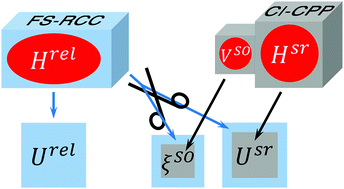
The spin-orbit (SO) interactions in low-lying electronic states of the LiM (M = Na, K, Rb, Cs) molecular series are studied through ab initio calculations of potential energy curves and SO coupling matrix elements as functions of the interatomic distance, R. Two different approaches are employed: (a) the Fock-space relativistic coupled-cluster calculations (FS-RCC) which directly yield full relativistic energies, Urel(R); the SO coupling functions, ξso(R), are extracted a posteriori through projecting scalar-relativistic wave functions onto the subspaces spanned by their full-relativistic counterparts; (b) the evaluation of the scalar-relativistic electronic energies, Usr(R), and relevant ξso(R) functions using the configuration interaction method with core-valence correlation accounted for using core polarization potentials (CI-CPP). The SO-free potentials and SO coupling functions obtained within the framework of both approaches are in good agreement with each other and their prior theoretical and empirical counterparts.
中文翻译:

LiM(M = Na,K,Rb,Cs)分子中自旋轨道耦合效应的第一个原理研究。
通过从头算计算势能曲线和SO耦合矩阵元素作为原子间的函数,研究了LiM(M = Na,K,Rb,Cs)分子序列的低电子态的自旋轨道(SO)相互作用采用两种不同的方法:(a)直接产生全部相对论能量的福克空间相对论耦合簇计算(FS-RCC);SO耦合函数ξso(R)是通过将标量相对论波函数投影到其完全相对论对应子空间上的子空间而提取出来的。(b)使用配置相互作用方法对标量相对论电子能量Usr(R)和相关的ξso(R)函数进行评估,并使用核极化价(CI-CPP)进行核价关联。
更新日期:2020-01-15
中文翻译:
LiM(M = Na,K,Rb,Cs)分子中自旋轨道耦合效应的第一个原理研究。
通过从头算计算势能曲线和SO耦合矩阵元素作为原子间的函数,研究了LiM(M = Na,K,Rb,Cs)分子序列的低电子态的自旋轨道(SO)相互作用采用两种不同的方法:(a)直接产生全部相对论能量的福克空间相对论耦合簇计算(FS-RCC);SO耦合函数ξso(R)是通过将标量相对论波函数投影到其完全相对论对应子空间上的子空间而提取出来的。(b)使用配置相互作用方法对标量相对论电子能量Usr(R)和相关的ξso(R)函数进行评估,并使用核极化价(CI-CPP)进行核价关联。


























 京公网安备 11010802027423号
京公网安备 11010802027423号