当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Bioinformatics Analysis of Metabolomics Data Unveils Association of Metabolic Signatures with Methylation in Breast Cancer.
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2019-12-30 , DOI: 10.1021/acs.jproteome.9b00755 Fadhl M Alakwaa 1 , Masha G Savelieff 1
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2019-12-30 , DOI: 10.1021/acs.jproteome.9b00755 Fadhl M Alakwaa 1 , Masha G Savelieff 1
Affiliation
|
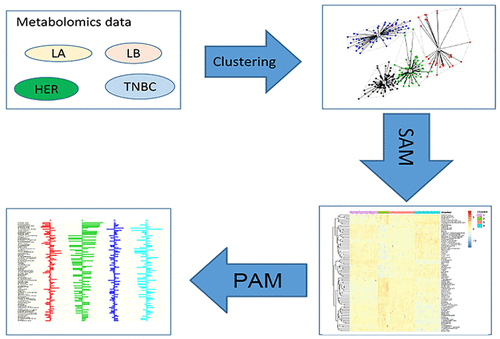
Breast cancer (BC) contributes the highest global cancer mortality in women. BC tumors are highly heterogeneous, so subtyping by cell-surface markers is inadequate. Omics-driven tumor stratification is urgently needed to better understand BC and tailor therapies for personalized medicine. We used unsupervised k-means and partition around medoids (pam) to cluster metabolomics data from two data sets. The first comprised 271 BC tumors (data set 1) that were estrogen receptor (ER) positive (ER+, n = 204) or negative (ER–, n = 67) with 162 identified and validated metabolites. The second data set contained 67 BC samples (data set 2; ER+, n = 33; ER–, n = 34) and 352 known metabolites. Significance Analysis of Microarrays (SAM) was used to identify the most significant metabolites among these clusters, which were then reassigned into new clusters using prediction analysis of microarrays (PAM). Generally, metabolome-defined BC subtypes identified from either data set 1 or data set 2 were different from the well-known receptor- or transcriptome-defined subtypes. Metabolomics-directed clustering of data set 2 identified distinctive BC tumors characterized by metabolome profiles that associated with DNA methylation (p-value = 0.000 048, χ2 test). Pathway analysis of cluster metabolites revealed that nitrogen metabolism and aminoacyl-tRNA biosynthesis were highly related to BC subtyping. The pipeline may be run from GitHub: https://github.com/FADHLyemen/Metabolomics_signature. Our proposed bioinformatics pipeline analyzed metabolomics data from BC tumors, revealing clusters characterized by unique metabolic signatures that may potentially stratify BC patients and tailor precision treatment.
中文翻译:

代谢组学数据的生物信息学分析揭示了乳腺癌中代谢特征与甲基化的关联。
乳腺癌(BC)是女性罹患全球癌症的最高死亡率。BC肿瘤高度异质,因此通过细胞表面标记物进行亚型分析是不够的。迫切需要组学驱动的肿瘤分层,以更好地了解BC并为个性化医学定制疗法。我们使用无监督的k均值和围绕类固醇(pam)的分区对来自两个数据集的代谢组学数据进行聚类。第一个包括271例BC肿瘤(数据集1),其中雌激素受体(ER)阳性(ER +,n = 204)或阴性(ER–,n = 67),其中有162种经鉴定和验证的代谢物。第二个数据集包含67个BC样本(数据集2; ER +,n = 33; ER–,n= 34)和352种已知代谢物。使用微阵列的重要性分析(SAM)来识别这些簇中最重要的代谢物,然后使用微阵列的预测分析(PAM)将其重新分配到新的簇中。通常,从数据集1或数据集2鉴定的代谢组定义的BC亚型与众所周知的受体或转录组定义的亚型不同。数据的代谢组学定向聚类组2个识别独特BC肿瘤特征在于代谢概况,与DNA甲基化相关(p -值= 0.000 048,χ 2测试)。簇代谢产物的途径分析表明,氮代谢和氨酰基-tRNA的生物合成与BC亚型高度相关。该管道可以从GitHub运行:https://github.com/FADHLyemen/Metabolomics_signature。我们提议的生物信息学渠道分析了来自BC肿瘤的代谢组学数据,揭示了以独特的代谢特征为特征的簇,这些特征可能潜在地使BC患者分层并调整精确治疗。
更新日期:2019-12-30
中文翻译:
代谢组学数据的生物信息学分析揭示了乳腺癌中代谢特征与甲基化的关联。
乳腺癌(BC)是女性罹患全球癌症的最高死亡率。BC肿瘤高度异质,因此通过细胞表面标记物进行亚型分析是不够的。迫切需要组学驱动的肿瘤分层,以更好地了解BC并为个性化医学定制疗法。我们使用无监督的k均值和围绕类固醇(pam)的分区对来自两个数据集的代谢组学数据进行聚类。第一个包括271例BC肿瘤(数据集1),其中雌激素受体(ER)阳性(ER +,n = 204)或阴性(ER–,n = 67),其中有162种经鉴定和验证的代谢物。第二个数据集包含67个BC样本(数据集2; ER +,n = 33; ER–,n= 34)和352种已知代谢物。使用微阵列的重要性分析(SAM)来识别这些簇中最重要的代谢物,然后使用微阵列的预测分析(PAM)将其重新分配到新的簇中。通常,从数据集1或数据集2鉴定的代谢组定义的BC亚型与众所周知的受体或转录组定义的亚型不同。数据的代谢组学定向聚类组2个识别独特BC肿瘤特征在于代谢概况,与DNA甲基化相关(p -值= 0.000 048,χ 2测试)。簇代谢产物的途径分析表明,氮代谢和氨酰基-tRNA的生物合成与BC亚型高度相关。该管道可以从GitHub运行:https://github.com/FADHLyemen/Metabolomics_signature。我们提议的生物信息学渠道分析了来自BC肿瘤的代谢组学数据,揭示了以独特的代谢特征为特征的簇,这些特征可能潜在地使BC患者分层并调整精确治疗。








































 京公网安备 11010802027423号
京公网安备 11010802027423号