当前位置:
X-MOL 学术
›
Chem. Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Artificial Intelligence to Accelerate the Discovery of N2 Electroreduction Catalysts
Chemistry of Materials ( IF 7.2 ) Pub Date : 2020-01-13 , DOI: 10.1021/acs.chemmater.9b03686 Myungjoon Kim 1 , Byung Chul Yeo 1 , Youngtae Park 2 , Hyuck Mo Lee 2 , Sang Soo Han 1 , Donghun Kim 1
Chemistry of Materials ( IF 7.2 ) Pub Date : 2020-01-13 , DOI: 10.1021/acs.chemmater.9b03686 Myungjoon Kim 1 , Byung Chul Yeo 1 , Youngtae Park 2 , Hyuck Mo Lee 2 , Sang Soo Han 1 , Donghun Kim 1
Affiliation
|
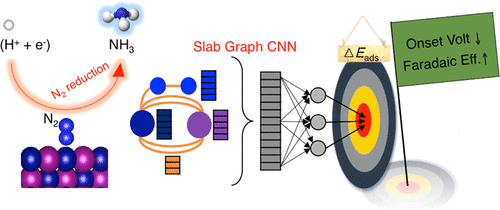
The development of catalysts for the electrochemical N2 reduction reaction (NRR) with a low limiting potential and high Faradaic efficiency is highly desirable but remains challenging. Here, to achieve acceleration, we develop and report a slab graph convolutional neural network (SGCNN), an accurate and flexible machine learning (ML) model that is suited for probing surface reactions in catalysis. With a self-accumulated database of 3040 surface calculations at the density-functional-theory (DFT) level, SGCNN predicted the binding energies, ranging over 8 eV, of five key adsorbates (H, N2, N2H, NH, NH2) related to NRR performance with a mean absolute error (MAE) of only 0.23 eV. SGCNN only requires the low-level inputs of elemental properties available in the periodic table of elements and connectivity information of constituent atoms; thus, accelerations can be realized. Via a combined process of SGCNN-driven predictions and DFT verifications, four novel catalysts in the L12 crystal space, including V3Ir(111), Tc3Hf(111), V3Ni(111), and Tc3Ta(111), are proposed as stable candidates that likely exhibit both a lower limiting potential and higher Faradaic efficiency in the NRR, relative to the reference Mo(110). The ML work combined with a statistical data analysis reveals that catalytic surfaces with an average d-orbital occupation between 4 and 6 could lower the limiting potential and potentially overcome the scaling relation in the NRR.
中文翻译:

人工智能促进N 2电还原催化剂的发现
迫切需要开发具有低极限电位和高法拉第效率的用于电化学N 2还原反应(NRR)的催化剂,但仍具有挑战性。在这里,为了实现加速,我们开发并报告了平板图卷积神经网络(SGCNN),这是一种适用于探测催化中的表面反应的准确而灵活的机器学习(ML)模型。利用在密度泛函理论(DFT)级别的3040个表面计算的自积累数据库,SGCNN预测了5种关键吸附物(H,N 2,N 2 H,NH,NH 2个)与NRR性能有关,平均绝对误差(MAE)仅0.23 eV。SGCNN仅需要元素周期表中可用的元素属性的低级输入以及组成原子的连接性信息;因此,可以实现加速。通过SGCNN驱动的预测和DFT验证的组合过程,L1 2晶体空间中的四种新型催化剂,包括V 3 Ir(111),Tc 3 Hf(111),V 3 Ni(111)和Tc 3提出Ta(111)是稳定的候选物,相对于参考Mo(110),它在NRR中可能同时显示出较低的极限电势和较高的法拉第效率。ML工作与统计数据分析相结合,发现平均d轨道占据在4到6之间的催化表面可以降低极限电位,并有可能克服NRR中的比例关系。
更新日期:2020-01-13
中文翻译:
人工智能促进N 2电还原催化剂的发现
迫切需要开发具有低极限电位和高法拉第效率的用于电化学N 2还原反应(NRR)的催化剂,但仍具有挑战性。在这里,为了实现加速,我们开发并报告了平板图卷积神经网络(SGCNN),这是一种适用于探测催化中的表面反应的准确而灵活的机器学习(ML)模型。利用在密度泛函理论(DFT)级别的3040个表面计算的自积累数据库,SGCNN预测了5种关键吸附物(H,N 2,N 2 H,NH,NH 2个)与NRR性能有关,平均绝对误差(MAE)仅0.23 eV。SGCNN仅需要元素周期表中可用的元素属性的低级输入以及组成原子的连接性信息;因此,可以实现加速。通过SGCNN驱动的预测和DFT验证的组合过程,L1 2晶体空间中的四种新型催化剂,包括V 3 Ir(111),Tc 3 Hf(111),V 3 Ni(111)和Tc 3提出Ta(111)是稳定的候选物,相对于参考Mo(110),它在NRR中可能同时显示出较低的极限电势和较高的法拉第效率。ML工作与统计数据分析相结合,发现平均d轨道占据在4到6之间的催化表面可以降低极限电位,并有可能克服NRR中的比例关系。










































 京公网安备 11010802027423号
京公网安备 11010802027423号