Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
The structure of Plasmodium falciparum hydroxymethyldihydropterin pyrophosphokinase-dihydropteroate synthase reveals the basis of sulfa resistance.
The FEBS Journal ( IF 5.5 ) Pub Date : 2019-12-28 , DOI: 10.1111/febs.15196 Penchit Chitnumsub 1 , Aritsara Jaruwat 1 , Yuwadee Talawanich 1 , Krittikar Noytanom 1 , Benjamas Liwnaree 1 , Sinothai Poen 1 , Yongyuth Yuthavong 1
The FEBS Journal ( IF 5.5 ) Pub Date : 2019-12-28 , DOI: 10.1111/febs.15196 Penchit Chitnumsub 1 , Aritsara Jaruwat 1 , Yuwadee Talawanich 1 , Krittikar Noytanom 1 , Benjamas Liwnaree 1 , Sinothai Poen 1 , Yongyuth Yuthavong 1
Affiliation
|
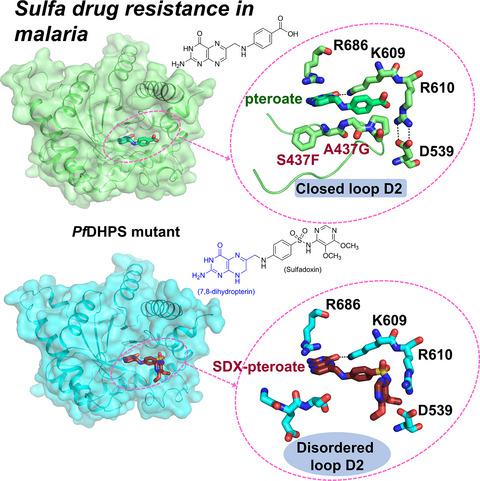
The clinical efficacy of sulfa drugs as antimalarials has declined owing to the evolution of resistance in Plasmodium falciparum (Pf ) malaria parasites. In order to understand the basis of this resistance and to design more effective antimalarials, we have solved 13 structures of the bifunctional enzyme 6‐hydroxymethyl‐7,8‐dihydropterin pyrophosphokinase (HPPK)–dihydropteroate synthase (DHPS) from wild‐type (WT) P. falciparum and sulfa‐resistant mutants, both as apoenzyme and as complexes with pteroate (PTA) and sulfa derivatives. The structures of these complexes show that PTA, which effectively inhibits both the WT and mutants, stays in active sites without steric constraint. In contrast, parts of the sulfa compounds situated outside of the substrate envelope are in the vicinity of the resistance mutations. Steric conflict between compound and mutant residue along with increased flexibility of loop D2 in the mutants can account for the reduced compound binding affinity to the mutants. Kinetic data show that the mutants have enhanced enzyme activity compared with the WT. These Pf DHPS structural insights are critical for the design of novel, substrate envelope–compliant DHPS inhibitors that are less vulnerable to resistance mutations.
中文翻译:

恶性疟原虫羟甲基二氢蝶呤焦磷酸激酶-二氢蝶呤合酶的结构揭示了耐磺胺性的基础。
由于恶性疟原虫(Pf)疟疾寄生虫产生了耐药性,因此磺胺类药物作为抗疟药的临床疗效有所下降。为了了解这种抗药性的基础并设计出更有效的抗疟药,我们从野生型(WT)中解决了13种双功能酶6-羟甲基-7,8-二氢蝶呤焦磷酸激酶(HPPK)-二氢蝶呤合酶(DHPS)的结构)恶性疟原虫和抗磺胺酸的突变体,既是脱辅酶,又是与蝶呤(PTA)和磺胺衍生物的复合物。这些复合物的结构表明,有效抑制野生型和突变体的PTA停留在没有空间限制的活性位点。相反,位于底物包膜外部的部分磺胺化合物在抗性突变的附近。化合物和突变体残基之间的立体冲突以及突变体中环D2的灵活性提高,可以解释化合物与突变体的结合亲和力降低。动力学数据表明,与野生型相比,突变体具有增强的酶活性。这些PfDHPS的结构见解对于设计新颖,与底物包膜相符的DHPS抑制剂至关重要,该抑制剂不易产生抗药性突变。
更新日期:2019-12-28
中文翻译:
恶性疟原虫羟甲基二氢蝶呤焦磷酸激酶-二氢蝶呤合酶的结构揭示了耐磺胺性的基础。
由于恶性疟原虫(Pf)疟疾寄生虫产生了耐药性,因此磺胺类药物作为抗疟药的临床疗效有所下降。为了了解这种抗药性的基础并设计出更有效的抗疟药,我们从野生型(WT)中解决了13种双功能酶6-羟甲基-7,8-二氢蝶呤焦磷酸激酶(HPPK)-二氢蝶呤合酶(DHPS)的结构)恶性疟原虫和抗磺胺酸的突变体,既是脱辅酶,又是与蝶呤(PTA)和磺胺衍生物的复合物。这些复合物的结构表明,有效抑制野生型和突变体的PTA停留在没有空间限制的活性位点。相反,位于底物包膜外部的部分磺胺化合物在抗性突变的附近。化合物和突变体残基之间的立体冲突以及突变体中环D2的灵活性提高,可以解释化合物与突变体的结合亲和力降低。动力学数据表明,与野生型相比,突变体具有增强的酶活性。这些PfDHPS的结构见解对于设计新颖,与底物包膜相符的DHPS抑制剂至关重要,该抑制剂不易产生抗药性突变。










































 京公网安备 11010802027423号
京公网安备 11010802027423号