Redox Biology ( IF 10.7 ) Pub Date : 2019-12-28 , DOI: 10.1016/j.redox.2019.101415 Jin Wang 1 , Pingjun Zhu 1 , Ruibing Li 1 , Jun Ren 2 , Hao Zhou 3
|
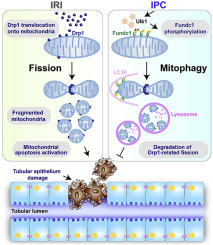
FUN14 domain-containing protein 1 (Fundc1)-dependent mitophagy, mainly activated by ischemic/hypoxic preconditioning, benefits acute myocardial reperfusion injury and chronic metabolic syndrome via sustaining mitochondrial homeostasis. Mitochondrial fission plays a pathogenic role in ischemic acute kidney injury (AKI) through perturbation of mitochondrial quality and activation of mitochondrial apoptosis. The aim of our study was to explore the role of Fundc1 mitophagy in ischemia preconditioning (IPC)-mediated renoprotection. Proximal tubule-specific Fundc1 knockout (Fundc1PTKO) mice were subjected to ischemia reperfusion injury (IRI) and IPC prior to assessment of renal function, mitophagy, mitochondrial quality control, and Drp1-related mitochondrial fission. Following exposure to IPC, Fundc1 mitophagy was activated through post-transcriptional phosphorylation at Ser17. Interestingly, IRI-mediated renal injury, inflammation, and tubule cell death were mitigated by IPC whereas proximal tubule-specific Fundc1 knockout (Fundc1PTKO) mice abolished IPC-offered renoprotection. Mechanistically, IRI-evoked mitochondrial damage was improved by IPC whereas Fundc1 deficiency provoked mitochondrial abnormality, manifested by impaired mitochondrial quality and hyperactivated Drp1-dependent mitochondrial fission. Interestingly, Fundc1 deficiency-associated mitochondrial dysfunction was reversed by pharmacological inhibition of mitochondrial fission. In vivo, Fundc1 deletion-caused renal injury, severe pro-inflammatory response, and tubule cell death could be nullified by way of knockout Drp1 on Fundc1PTKO background. Finally, we also revealed that IPC triggered Fundc1 mitophagy activation through UNC-51-like kinase 1 (Ulk1) and Ulk1 ablation interrupted IPC-mediated Fundc1 activation and thus attenuated IPC-induced renoprotection. Fundc1 mitophagy, primarily driven by IPC, confers resistance to AKI through reconciliation of mitochondrial fission, implicating the therapeutic potential of targeting mitochondrial homeostasis for AKI.
中文翻译:
通过抑制Drp1介导的线粒体裂变,Fundc1依赖性线粒体对缺血性AKI中缺血预适应赋予的肾脏保护是必不可少的。
主要通过缺血/低氧预处理激活的含FUN14域蛋白1(Fundc1)依赖性线粒体通过维持线粒体体内稳态,有利于急性心肌再灌注损伤和慢性代谢综合征。线粒体裂变通过扰动线粒体质量和激活线粒体凋亡在缺血性急性肾损伤(AKI)中发挥致病作用。我们研究的目的是探讨Fundc1线粒体在缺血预处理(IPC)介导的肾脏保护中的作用。近端小管特异性Fundc1敲除(Fundc1 PTKO)小鼠在评估肾功能,线粒体,线粒体质量控制和Drp1相关的线粒体裂变之前经历了缺血再灌注损伤(IRI)和IPC。暴露于IPC之后,Ser17的转录后磷酸化激活了Fundc1的细胞吞噬作用。有趣的是,IPC减轻了IRI介导的肾损伤,炎症和肾小管细胞死亡,而近端肾小管特异性Fundc1基因敲除(Fundc1PTKO)小鼠取消了IPC提供的肾脏保护。从机理上讲,IPC改善了IRI引起的线粒体损伤,而Fundc1缺乏引起线粒体异常,表现为线粒体质量受损和过度激活的Drp1依赖性线粒体裂变。有趣的是,通过药理学抑制线粒体裂变,逆转了与Fundc1缺乏相关的线粒体功能障碍。在体内,可通过在Fundc1PTKO背景上敲除Drp1来消除Fundc1缺失引起的肾损伤,严重的促炎反应和肾小管细胞死亡。最后,我们还揭示了IPC通过UNC-51样激酶1(Ulk1)和Ulk1触发了Fundc1线粒体激活。消融中断了IPC介导的Fundc1激活,从而减弱了IPC诱导的肾保护作用。主要由IPC驱动的Fundc1线粒体吞噬通过调节线粒体裂变赋予对AKI的抗药性,这暗示了针对AKI靶向线粒体稳态的治疗潜力。










































 京公网安备 11010802027423号
京公网安备 11010802027423号