当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Characterizing Protein-Ligand Binding Using Atomistic Simulation and Machine Learning: Application to Drug Resistance in HIV-1 Protease.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2019-12-26 , DOI: 10.1021/acs.jctc.9b00781 Troy W Whitfield 1, 2 , Debra A Ragland 3 , Konstantin B Zeldovich 2 , Celia A Schiffer 3
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2019-12-26 , DOI: 10.1021/acs.jctc.9b00781 Troy W Whitfield 1, 2 , Debra A Ragland 3 , Konstantin B Zeldovich 2 , Celia A Schiffer 3
Affiliation
|
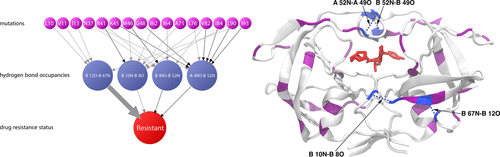
Over the past several decades, atomistic simulations of biomolecules, whether carried out using molecular dynamics or Monte Carlo techniques, have provided detailed insights into their function. Comparing the results of such simulations for a few closely related systems has guided our understanding of the mechanisms by which changes such as ligand binding or mutation can alter the function. The general problem of detecting and interpreting such mechanisms from simulations of many related systems, however, remains a challenge. This problem is addressed here by applying supervised and unsupervised machine learning techniques to a variety of thermodynamic observables extracted from molecular dynamics simulations of different systems. As an important test case, these methods are applied to understand the evasion by human immunodeficiency virus type-1 (HIV-1) protease of darunavir, a potent inhibitor to which resistance can develop via the simultaneous mutation of multiple amino acids. Complex mutational patterns have been observed among resistant strains, presenting a challenge to developing a mechanistic picture of resistance in the protease. In order to dissect these patterns and gain mechanistic insight into the role of specific mutations, molecular dynamics simulations were carried out on a collection of HIV-1 protease variants, chosen to include highly resistant strains and susceptible controls, in complex with darunavir. Using a machine learning approach that takes advantage of the hierarchical nature in the relationships among the sequence, structure, and function, an integrative analysis of these trajectories reveals key details of the resistance mechanism, including changes in the protein structure, hydrogen bonding, and protein-ligand contacts.
中文翻译:

使用原子模拟和机器学习表征蛋白质-配体结合:在HIV-1蛋白酶的耐药性中的应用。
在过去的几十年中,无论是使用分子动力学还是蒙特卡洛技术进行的生物分子的原子模拟,都为它们的功能提供了详细的见识。比较一些紧密相关系统的模拟结果,可以指导我们理解配体结合或突变等改变可以改变功能的机制。然而,从许多相关系统的仿真中检测和解释这种机制的一般问题仍然是一个挑战。通过将有监督的和无监督的机器学习技术应用于从不同系统的分子动力学模拟中提取的各种热力学可观测值,可以解决此问题。作为重要的测试案例,这些方法用于了解人类免疫缺陷病毒1型(HIV-1)蛋白酶对达那韦的逃避作用,达那那韦是一种有效的抑制剂,通过同时突变多个氨基酸可产生耐药性。已在耐药菌株中观察到复杂的突变模式,这对发展蛋白酶中耐药机制的研究提出了挑战。为了剖析这些模式并获得对特定突变作用的机械理解,对一系列与darunavir结合的HIV-1蛋白酶变异体进行了分子动力学模拟,这些变异体包括高抗性菌株和易感对照。使用在序列,结构和功能之间的关系中利用分层性质的机器学习方法,
更新日期:2020-01-17
中文翻译:
使用原子模拟和机器学习表征蛋白质-配体结合:在HIV-1蛋白酶的耐药性中的应用。
在过去的几十年中,无论是使用分子动力学还是蒙特卡洛技术进行的生物分子的原子模拟,都为它们的功能提供了详细的见识。比较一些紧密相关系统的模拟结果,可以指导我们理解配体结合或突变等改变可以改变功能的机制。然而,从许多相关系统的仿真中检测和解释这种机制的一般问题仍然是一个挑战。通过将有监督的和无监督的机器学习技术应用于从不同系统的分子动力学模拟中提取的各种热力学可观测值,可以解决此问题。作为重要的测试案例,这些方法用于了解人类免疫缺陷病毒1型(HIV-1)蛋白酶对达那韦的逃避作用,达那那韦是一种有效的抑制剂,通过同时突变多个氨基酸可产生耐药性。已在耐药菌株中观察到复杂的突变模式,这对发展蛋白酶中耐药机制的研究提出了挑战。为了剖析这些模式并获得对特定突变作用的机械理解,对一系列与darunavir结合的HIV-1蛋白酶变异体进行了分子动力学模拟,这些变异体包括高抗性菌株和易感对照。使用在序列,结构和功能之间的关系中利用分层性质的机器学习方法,










































 京公网安备 11010802027423号
京公网安备 11010802027423号