当前位置:
X-MOL 学术
›
Int. J. Quantum Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Constructing spin-adiabatic states for the modeling of spin-crossing reactions. I. A shared-orbital implementation
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2019-12-24 , DOI: 10.1002/qua.26123 Yunwen Tao 1 , Zheng Pei 2 , Nicole Bellonzi 3 , Yuezhi Mao 4 , Zhu Zou 1 , Wanzhen Liang 2 , Zhibo Yang 1 , Yihan Shao 1
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2019-12-24 , DOI: 10.1002/qua.26123 Yunwen Tao 1 , Zheng Pei 2 , Nicole Bellonzi 3 , Yuezhi Mao 4 , Zhu Zou 1 , Wanzhen Liang 2 , Zhibo Yang 1 , Yihan Shao 1
Affiliation
|
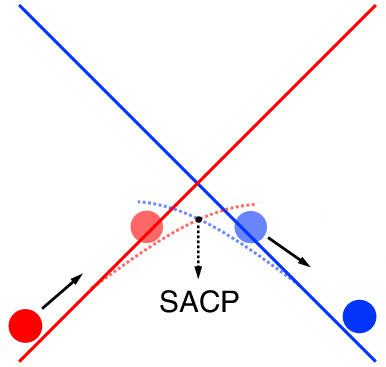
In the modeling of spin-crossing reactions, it has become popular to directly explore the spin-adiabatic surfaces. Specifically, through constructing spin-adiabatic states from a two-state Hamiltonian (with spin-orbit coupling matrix elements) at each geometry, one can readily employ advanced geometry optimization algorithms to acquire a "transition state" structure, where the spin crossing occurs. In this work, we report the implementation of a fully-variational spin-adiabatic approach based on Kohn-Sham density functional theory spin states (sharing the same set of molecular orbitals) and the Breit-Pauli one-electron spin-orbit operator. For three model spin-crossing reactions [predissociation of N2O, singlet-triplet conversion in CH2, and CO addition to Fe(CO)4], the spin-crossing points were obtained. Our results also indicated the Breit-Pauli one-electron spin-orbit coupling can vary significantly along the reaction pathway on the spin-adiabatic energy surface. On the other hand, due to the restriction that low-spin and high-spin states share the same set of molecular orbitals, the acquired spin-adiabatic energy surface shows a cusp (i.e. a first-order discontinuity) at the crossing point, which prevents the use of standard geometry optimization algorithms to pinpoint the crossing point. An extension with this restriction removed is being developed to achieve the smoothness of spin-adiabatic surfaces.
中文翻译:

构建用于自旋交叉反应建模的自旋绝热状态。I. 共享轨道实现
在自旋交叉反应的建模中,直接探索自旋绝热表面已经变得流行。具体来说,通过在每个几何结构上从二态哈密顿量(具有自旋轨道耦合矩阵元素)构建自旋绝热态,人们可以很容易地采用先进的几何优化算法来获得“过渡态”结构,其中发生自旋交叉。在这项工作中,我们报告了基于 Kohn-Sham 密度泛函理论自旋态(共享同一组分子轨道)和 Breit-Pauli 单电子自旋轨道算子的全变分自旋-绝热方法的实现。对于三个模型自旋交叉反应 [N2O 的预解离、CH2 中的单线态-三线态转化和 CO 添加到 Fe(CO)4],获得了自旋交叉点。我们的结果还表明 Breit-Pauli 单电子自旋轨道耦合可以沿着自旋绝热能量表面上的反应路径显着变化。另一方面,由于低自旋态和高自旋态共享同一组分子轨道的限制,获得的自旋-绝热能面在交点处显示出一个尖点(即一阶不连续性),即阻止使用标准几何优化算法来精确定位交叉点。正在开发消除此限制的扩展,以实现自旋绝热表面的平滑度。由于低自旋态和高自旋态共享同一组分子轨道的限制,获得的自旋绝热能面在交叉点处显示出尖峰(即一阶不连续性),这阻止了使用标准几何优化算法来确定交叉点。正在开发消除此限制的扩展,以实现自旋绝热表面的平滑度。由于低自旋态和高自旋态共享同一组分子轨道的限制,获得的自旋绝热能面在交叉点处显示出尖峰(即一阶不连续性),这阻止了使用标准几何优化算法来确定交叉点。正在开发消除此限制的扩展,以实现自旋绝热表面的平滑度。
更新日期:2019-12-24
中文翻译:
构建用于自旋交叉反应建模的自旋绝热状态。I. 共享轨道实现
在自旋交叉反应的建模中,直接探索自旋绝热表面已经变得流行。具体来说,通过在每个几何结构上从二态哈密顿量(具有自旋轨道耦合矩阵元素)构建自旋绝热态,人们可以很容易地采用先进的几何优化算法来获得“过渡态”结构,其中发生自旋交叉。在这项工作中,我们报告了基于 Kohn-Sham 密度泛函理论自旋态(共享同一组分子轨道)和 Breit-Pauli 单电子自旋轨道算子的全变分自旋-绝热方法的实现。对于三个模型自旋交叉反应 [N2O 的预解离、CH2 中的单线态-三线态转化和 CO 添加到 Fe(CO)4],获得了自旋交叉点。我们的结果还表明 Breit-Pauli 单电子自旋轨道耦合可以沿着自旋绝热能量表面上的反应路径显着变化。另一方面,由于低自旋态和高自旋态共享同一组分子轨道的限制,获得的自旋-绝热能面在交点处显示出一个尖点(即一阶不连续性),即阻止使用标准几何优化算法来精确定位交叉点。正在开发消除此限制的扩展,以实现自旋绝热表面的平滑度。由于低自旋态和高自旋态共享同一组分子轨道的限制,获得的自旋绝热能面在交叉点处显示出尖峰(即一阶不连续性),这阻止了使用标准几何优化算法来确定交叉点。正在开发消除此限制的扩展,以实现自旋绝热表面的平滑度。由于低自旋态和高自旋态共享同一组分子轨道的限制,获得的自旋绝热能面在交叉点处显示出尖峰(即一阶不连续性),这阻止了使用标准几何优化算法来确定交叉点。正在开发消除此限制的扩展,以实现自旋绝热表面的平滑度。










































 京公网安备 11010802027423号
京公网安备 11010802027423号