当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Systematic Modeling of log D7.4 Based on Ensemble Machine Learning, Group Contribution, and Matched Molecular Pair Analysis.
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-01-10 , DOI: 10.1021/acs.jcim.9b00718 Li Fu 1 , Lu Liu 1 , Zhi-Jiang Yang 1 , Pan Li 2 , Jun-Jie Ding 2 , Yong-Huan Yun 3 , Ai-Ping Lu 4 , Ting-Jun Hou 5 , Dong-Sheng Cao 1, 4
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-01-10 , DOI: 10.1021/acs.jcim.9b00718 Li Fu 1 , Lu Liu 1 , Zhi-Jiang Yang 1 , Pan Li 2 , Jun-Jie Ding 2 , Yong-Huan Yun 3 , Ai-Ping Lu 4 , Ting-Jun Hou 5 , Dong-Sheng Cao 1, 4
Affiliation
|
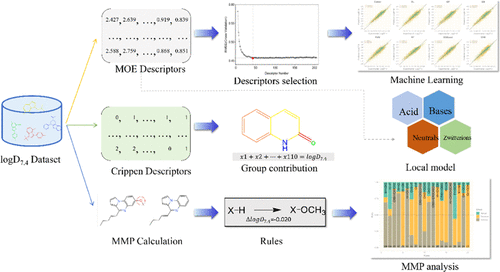
Lipophilicity, as evaluated by the n-octanol/buffer solution distribution coefficient at pH = 7.4 (log D7.4), is a major determinant of various absorption, distribution, metabolism, elimination, and toxicology (ADMET) parameters of drug candidates. In this study, we developed several quantitative structure-property relationship (QSPR) models to predict log D7.4 based on a large and structurally diverse data set. Eight popular machine learning algorithms were employed to build the prediction models with 43 molecular descriptors selected by a wrapper feature selection method. The results demonstrated that XGBoost yielded better prediction performance than any other single model (RT2 = 0.906 and RMSET = 0.395). Moreover, the consensus model from the top three models could continue to improve the prediction performance (RT2 = 0.922 and RMSET = 0.359). The robustness, reliability, and generalization ability of the models were strictly evaluated by the Y-randomization test and applicability domain analysis. Moreover, the group contribution model based on 110 atom types and the local models for different ionization states were also established and compared to the global models. The results demonstrated that the descriptor-based consensus model is superior to the group contribution method, and the local models have no advantage over the global models. Finally, matched molecular pair (MMP) analysis and descriptor importance analysis were performed to extract transformation rules and give some explanations related to log D7.4. In conclusion, we believe that the consensus model developed in this study can be used as a reliable and promising tool to evaluate log D7.4 in drug discovery.
中文翻译:

基于集成机器学习,基团贡献和匹配分子对分析的log D7.4系统建模。
亲脂性,通过在pH = 7.4时的正辛醇/缓冲溶液分配系数评估(对数D7.4),是候选药物的各种吸收,分布,代谢,消除和毒理学(ADMET)参数的主要决定因素。在这项研究中,我们开发了一些定量的结构-属性关系(QSPR)模型,以基于庞大且结构多样的数据集预测对数D7.4。采用了八种流行的机器学习算法,以通过包装特征选择方法选择的43个分子描述符来构建预测模型。结果表明,XGBoost的预测性能优于任何其他单个模型(RT2 = 0.906和RMSET = 0.395)。此外,来自前三个模型的共识模型可以继续提高预测性能(RT2 = 0.922和RMSET = 0.359)。模型的鲁棒性,可靠性和泛化能力通过Y随机检验和适用性域分析进行了严格评估。此外,还建立了基于110种原子类型的基团贡献模型和不同电离态的局部模型,并将其与全局模型进行了比较。结果表明,基于描述符的共识模型优于群体贡献方法,局部模型没有全局模型的优势。最后,进行了匹配分子对(MMP)分析和描述符重要性分析,以提取转化规则并给出与log D7.4相关的一些解释。总之,我们认为,本研究中开发的共识模型可以用作评估药物开发中log D7.4的可靠且有前途的工具。通过Y随机检验和适用性域分析,严格评估了模型的可靠性和泛化能力。此外,还建立了基于110种原子类型的基团贡献模型和不同电离态的局部模型,并将其与全局模型进行了比较。结果表明,基于描述符的共识模型优于群体贡献方法,局部模型没有全局模型的优势。最后,进行了匹配分子对(MMP)分析和描述符重要性分析,以提取转化规则并给出与log D7.4相关的一些解释。总之,我们认为,本研究中开发的共识模型可以用作评估药物开发中log D7.4的可靠且有前途的工具。通过Y随机检验和适用性域分析,严格评估了模型的可靠性和泛化能力。此外,还建立了基于110种原子类型的基团贡献模型和不同电离态的局部模型,并将其与全局模型进行了比较。结果表明,基于描述符的共识模型优于群体贡献方法,局部模型没有全局模型的优势。最后,进行了匹配分子对(MMP)分析和描述符重要性分析,以提取转化规则并给出与log D7.4相关的一些解释。总之,我们认为,本研究中开发的共识模型可以用作评估药物开发中log D7.4的可靠且有前途的工具。模型的泛化能力通过Y随机检验和适用性域分析进行严格评估。此外,还建立了基于110种原子类型的基团贡献模型和不同电离态的局部模型,并将其与全局模型进行了比较。结果表明,基于描述符的共识模型优于群体贡献方法,局部模型没有全局模型的优势。最后,进行了匹配分子对(MMP)分析和描述符重要性分析,以提取转化规则并给出与log D7.4相关的一些解释。总之,我们认为,本研究中开发的共识模型可以用作评估药物发现中log D7.4的可靠且有前途的工具。模型的泛化能力通过Y随机检验和适用性域分析进行严格评估。此外,还建立了基于110种原子类型的基团贡献模型和不同电离态的局部模型,并将其与全局模型进行了比较。结果表明,基于描述符的共识模型优于群体贡献方法,局部模型没有全局模型的优势。最后,进行了匹配分子对(MMP)分析和描述符重要性分析,以提取转化规则并给出与log D7.4相关的一些解释。总之,我们认为,本研究中开发的共识模型可以用作评估药物发现中log D7.4的可靠且有前途的工具。
更新日期:2020-01-10
中文翻译:
基于集成机器学习,基团贡献和匹配分子对分析的log D7.4系统建模。
亲脂性,通过在pH = 7.4时的正辛醇/缓冲溶液分配系数评估(对数D7.4),是候选药物的各种吸收,分布,代谢,消除和毒理学(ADMET)参数的主要决定因素。在这项研究中,我们开发了一些定量的结构-属性关系(QSPR)模型,以基于庞大且结构多样的数据集预测对数D7.4。采用了八种流行的机器学习算法,以通过包装特征选择方法选择的43个分子描述符来构建预测模型。结果表明,XGBoost的预测性能优于任何其他单个模型(RT2 = 0.906和RMSET = 0.395)。此外,来自前三个模型的共识模型可以继续提高预测性能(RT2 = 0.922和RMSET = 0.359)。模型的鲁棒性,可靠性和泛化能力通过Y随机检验和适用性域分析进行了严格评估。此外,还建立了基于110种原子类型的基团贡献模型和不同电离态的局部模型,并将其与全局模型进行了比较。结果表明,基于描述符的共识模型优于群体贡献方法,局部模型没有全局模型的优势。最后,进行了匹配分子对(MMP)分析和描述符重要性分析,以提取转化规则并给出与log D7.4相关的一些解释。总之,我们认为,本研究中开发的共识模型可以用作评估药物开发中log D7.4的可靠且有前途的工具。通过Y随机检验和适用性域分析,严格评估了模型的可靠性和泛化能力。此外,还建立了基于110种原子类型的基团贡献模型和不同电离态的局部模型,并将其与全局模型进行了比较。结果表明,基于描述符的共识模型优于群体贡献方法,局部模型没有全局模型的优势。最后,进行了匹配分子对(MMP)分析和描述符重要性分析,以提取转化规则并给出与log D7.4相关的一些解释。总之,我们认为,本研究中开发的共识模型可以用作评估药物开发中log D7.4的可靠且有前途的工具。通过Y随机检验和适用性域分析,严格评估了模型的可靠性和泛化能力。此外,还建立了基于110种原子类型的基团贡献模型和不同电离态的局部模型,并将其与全局模型进行了比较。结果表明,基于描述符的共识模型优于群体贡献方法,局部模型没有全局模型的优势。最后,进行了匹配分子对(MMP)分析和描述符重要性分析,以提取转化规则并给出与log D7.4相关的一些解释。总之,我们认为,本研究中开发的共识模型可以用作评估药物开发中log D7.4的可靠且有前途的工具。模型的泛化能力通过Y随机检验和适用性域分析进行严格评估。此外,还建立了基于110种原子类型的基团贡献模型和不同电离态的局部模型,并将其与全局模型进行了比较。结果表明,基于描述符的共识模型优于群体贡献方法,局部模型没有全局模型的优势。最后,进行了匹配分子对(MMP)分析和描述符重要性分析,以提取转化规则并给出与log D7.4相关的一些解释。总之,我们认为,本研究中开发的共识模型可以用作评估药物发现中log D7.4的可靠且有前途的工具。模型的泛化能力通过Y随机检验和适用性域分析进行严格评估。此外,还建立了基于110种原子类型的基团贡献模型和不同电离态的局部模型,并将其与全局模型进行了比较。结果表明,基于描述符的共识模型优于群体贡献方法,局部模型没有全局模型的优势。最后,进行了匹配分子对(MMP)分析和描述符重要性分析,以提取转化规则并给出与log D7.4相关的一些解释。总之,我们认为,本研究中开发的共识模型可以用作评估药物发现中log D7.4的可靠且有前途的工具。










































 京公网安备 11010802027423号
京公网安备 11010802027423号