当前位置:
X-MOL 学术
›
ChemPhysChem
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Alkali and Alkaline-Earth Cations in Complexes with Small Bioorganic Ligands: Ab Initio Benchmark Calculations and Bond Energy Decomposition.
ChemPhysChem ( IF 2.3 ) Pub Date : 2019-12-11 , DOI: 10.1002/cphc.201900877 Roberto López 1 , Natalia Díaz 2 , Dimas Suárez 2
ChemPhysChem ( IF 2.3 ) Pub Date : 2019-12-11 , DOI: 10.1002/cphc.201900877 Roberto López 1 , Natalia Díaz 2 , Dimas Suárez 2
Affiliation
|
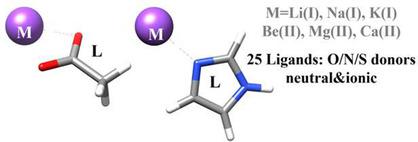
Herein, we report a computational database for the complexes of alkali [Li(I), Na(I), K(I)] and alkaline‐earth [Be(II), Mg(II) and Ca(II)] cations with 25 small ligands with varying charge and donor atoms (“O”, “N”, and “S”) that provides geometries and accurate bond energies useful to analyze metal‐ligand interactions in proteins and nucleic acids. The role of the ligand→metal charge transfer, the equilibrium bond distance, the electronegativity of the donor atom, the ligand polarizability, and the relative stability of the complexes are discussed in detail. The interacting quantum atoms (IQA) method is used to decompose the binding energy into electrostatic and quantum mechanical contributions. In addition, bond energies are also estimated by means of multipolar electrostatic calculations. No simple correlation exists between bond energies and structural/electronic descriptors unless the data are segregated by the type of ligand or metal. The electrostatic attraction of some molecules (H2O, NH3, CH3OH) towards the metal cations is well reproduced using their (unrelaxed) atomic multipoles, but the same comparison is much less satisfactory for other ligands (e. g. benzene, thiol/thiolate groups, etc.). Besides providing reference structures and bond energies, the database can contribute to validate molecular mechanics potentials capable of yielding a balanced description of alkali and alkaline‐earth metals binding to biomolecules.
中文翻译:

小型生物有机配体配合物中的碱和碱土阳离子:从头算基准计算和键能分解。
在这里,我们报告了碱金属[Li(I),Na(I),K(I)]和碱土金属[Be(II),Mg(II)和Ca(II)]阳离子与25个具有不同电荷和供体原子(“ O”,“ N”和“ S”)的小配体,可提供几何形状和准确的键能,可用于分析蛋白质和核酸中的金属配体相互作用。详细讨论了配体→金属电荷转移,平衡键距,供体原子的电负性,配体极化率和配合物的相对稳定性的作用。相互作用的量子原子(IQA)方法用于将结合能分解为静电和量子力学贡献。另外,还通过多极静电计算来估算键能。除非通过配体或金属的类型将数据分开,否则键能与结构/电子描述符之间不存在简单的关联。一些分子的静电引力(H使用它们的(非松弛)原子多极可很好地复制金属阳离子的2 O,NH 3,CH 3 OH),但是对于其他配体(例如苯,硫醇/硫醇盐等)而言,同样的比较效果却差强人意。除了提供参考结构和键能以外,该数据库还可以帮助验证分子力学潜力,从而能够对与生物分子结合的碱金属和碱土金属进行均衡的描述。
更新日期:2019-12-11
中文翻译:
小型生物有机配体配合物中的碱和碱土阳离子:从头算基准计算和键能分解。
在这里,我们报告了碱金属[Li(I),Na(I),K(I)]和碱土金属[Be(II),Mg(II)和Ca(II)]阳离子与25个具有不同电荷和供体原子(“ O”,“ N”和“ S”)的小配体,可提供几何形状和准确的键能,可用于分析蛋白质和核酸中的金属配体相互作用。详细讨论了配体→金属电荷转移,平衡键距,供体原子的电负性,配体极化率和配合物的相对稳定性的作用。相互作用的量子原子(IQA)方法用于将结合能分解为静电和量子力学贡献。另外,还通过多极静电计算来估算键能。除非通过配体或金属的类型将数据分开,否则键能与结构/电子描述符之间不存在简单的关联。一些分子的静电引力(H使用它们的(非松弛)原子多极可很好地复制金属阳离子的2 O,NH 3,CH 3 OH),但是对于其他配体(例如苯,硫醇/硫醇盐等)而言,同样的比较效果却差强人意。除了提供参考结构和键能以外,该数据库还可以帮助验证分子力学潜力,从而能够对与生物分子结合的碱金属和碱土金属进行均衡的描述。










































 京公网安备 11010802027423号
京公网安备 11010802027423号