当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
The hunt for reactive alkynes in bio-orthogonal click reactions: insights from mechanochemical and conceptual DFT calculations
Chemical Science ( IF 7.6 ) Pub Date : 2019/12/23 , DOI: 10.1039/c9sc04507d Tom Bettens 1, 2, 3, 4 , Mercedes Alonso 1, 2, 3, 4 , Paul Geerlings 1, 2, 3, 4 , Frank De Proft 1, 2, 3, 4
Chemical Science ( IF 7.6 ) Pub Date : 2019/12/23 , DOI: 10.1039/c9sc04507d Tom Bettens 1, 2, 3, 4 , Mercedes Alonso 1, 2, 3, 4 , Paul Geerlings 1, 2, 3, 4 , Frank De Proft 1, 2, 3, 4
Affiliation
|
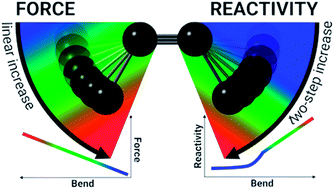
In our effort to implement the mechanical force used to activate single molecules in mechanochemistry in the context of conceptual density functional theory, we present a theoretical investigation of strained alkynes for rationalizing structural trends as well as the reactivity of cyclic alkynes that are of great importance in in vivo click reactions. The strain on the triple bond in cyclic alkynes is modeled by angular constraints in a 2-butyne fragment and the corresponding bending force is calculated by means of an extended COGEF (constrained geometries simulate external forces) model. In general, the force required to bend the triple bond is smaller with electron-withdrawing groups on the propargylic C-atom, which elegantly results in smaller angles around the triple bond in cyclic alkynes with such substitution pattern. By means of conceptual DFT descriptors, the electrophilic and nucleophilic character of bent triple bonds was investigated revealing moderate activation for small distortions from the linear geometry (0° to 15°) and a drastically more reactive π-space if the triple bond is bent further. This analysis of the intrinsic reactivity of the triple bond is in line with experimental observations, explaining the reactive nature of cyclooctynes and cycloheptynes, whereas larger cyclic systems do not drastically activate the triple bond.
中文翻译:

在生物正交点击反应中寻找反应性炔烃:机械化学和概念DFT计算的见解
为了在概念密度泛函理论的背景下实现机械化学中用于激活单分子的机械力,我们对张力炔烃进行了理论研究,以合理化结构趋势以及对环状炔烃的反应性,这在化学反应中非常重要。体内点击反应。通过2-丁炔片段中的角度约束对环状炔烃中三键上的应变进行建模,并通过扩展的COGEF(受约束的几何形状模拟外力)模型来计算相应的弯曲力。通常,在炔丙基碳原子上具有吸电子基团时,弯曲三键所需的力较小,这优雅地导致了具有这种取代方式的环状炔烃中三键周围的夹角较小。通过概念性DFT描述子,研究了弯曲的三键的亲电和亲核特性,揭示了线性几何结构(0°至15°)的小畸变具有适度的活化作用,如果三键进一步弯曲,则π-空间的反应性大大增强。
更新日期:2020-02-13
中文翻译:
在生物正交点击反应中寻找反应性炔烃:机械化学和概念DFT计算的见解
为了在概念密度泛函理论的背景下实现机械化学中用于激活单分子的机械力,我们对张力炔烃进行了理论研究,以合理化结构趋势以及对环状炔烃的反应性,这在化学反应中非常重要。体内点击反应。通过2-丁炔片段中的角度约束对环状炔烃中三键上的应变进行建模,并通过扩展的COGEF(受约束的几何形状模拟外力)模型来计算相应的弯曲力。通常,在炔丙基碳原子上具有吸电子基团时,弯曲三键所需的力较小,这优雅地导致了具有这种取代方式的环状炔烃中三键周围的夹角较小。通过概念性DFT描述子,研究了弯曲的三键的亲电和亲核特性,揭示了线性几何结构(0°至15°)的小畸变具有适度的活化作用,如果三键进一步弯曲,则π-空间的反应性大大增强。










































 京公网安备 11010802027423号
京公网安备 11010802027423号