当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Ensemble-Based Molecular Simulation of Chemical Reactions under Vibrational Nonequilibrium.
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2019-12-30 , DOI: 10.1021/acs.jpclett.9b03356 Kristof M Bal 1 , Annemie Bogaerts 1 , Erik C Neyts 1
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2019-12-30 , DOI: 10.1021/acs.jpclett.9b03356 Kristof M Bal 1 , Annemie Bogaerts 1 , Erik C Neyts 1
Affiliation
|
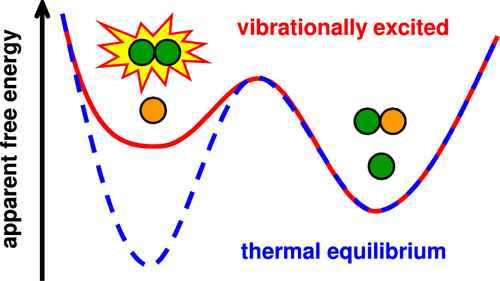
We present an approach to incorporate the effect of vibrational nonequilibrium in molecular dynamics (MD) simulations. A perturbed canonical ensemble, in which selected modes are excited to higher temperature while all others remain equilibrated at low temperature, is simulated by applying a specifically tailored bias potential. Our method can be readily applied to any (classical or quantum mechanical) MD setup at virtually no additional computational cost and allows the study of reactions of vibrationally excited molecules in nonequilibrium environments such as plasmas. In combination with enhanced sampling methods, the vibrational efficacy and mode selectivity of vibrationally stimulated reactions can then be quantified in terms of chemically relevant observables, such as reaction rates and apparent free energy barriers. We first validate our method for the prototypical hydrogen exchange reaction and then show how it can capture the effect of vibrational excitation on a symmetric SN2 reaction and radical addition on CO2.
中文翻译:

振动非平衡下基于集合体的化学反应分子模拟。
我们提出了一种在分子动力学(MD)模拟中纳入振动非平衡效应的方法。通过应用专门定制的偏置电势,可以模拟扰动的典范合奏,在该合奏中,选定的模式被激发到更高的温度,而所有其他模式在低温下保持平衡。我们的方法可以很容易地应用于任何(经典或量子力学)MD装置,而实际上无需额外的计算成本,并且可以研究在非平衡环境(例如等离子体)中振动激发的分子的反应。结合增强的采样方法,然后可以根据化学相关的可观测值(例如反应速率和表观自由能垒)来量化振动激发反应的振动效率和模式选择性。
更新日期:2019-12-30
中文翻译:
振动非平衡下基于集合体的化学反应分子模拟。
我们提出了一种在分子动力学(MD)模拟中纳入振动非平衡效应的方法。通过应用专门定制的偏置电势,可以模拟扰动的典范合奏,在该合奏中,选定的模式被激发到更高的温度,而所有其他模式在低温下保持平衡。我们的方法可以很容易地应用于任何(经典或量子力学)MD装置,而实际上无需额外的计算成本,并且可以研究在非平衡环境(例如等离子体)中振动激发的分子的反应。结合增强的采样方法,然后可以根据化学相关的可观测值(例如反应速率和表观自由能垒)来量化振动激发反应的振动效率和模式选择性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号