当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Ab Initio Many-Body Perturbation Theory Calculations of the Electronic and Optical Properties of Cyclometalated Ir(III) Complexes.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-01-06 , DOI: 10.1021/acs.jctc.9b00763 Marco Cazzaniga 1 , Fausto Cargnoni 1 , Marta Penconi 1 , Alberto Bossi 1 , Davide Ceresoli 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2020-01-06 , DOI: 10.1021/acs.jctc.9b00763 Marco Cazzaniga 1 , Fausto Cargnoni 1 , Marta Penconi 1 , Alberto Bossi 1 , Davide Ceresoli 1
Affiliation
|
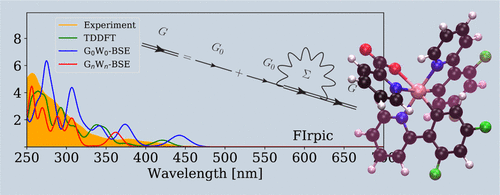
Cyclometalated Ir(III) compounds are the preferred choice as organic emitters in organic light-emitting diodes. In practice, the presence of the transition metal surrounded by carefully designed ligands allows fine-tuning of the emission frequency as well as good efficiency of the device. To support the development of new compounds, experimental measurements are generally compared with absorption and emission spectra obtained from ab initio calculations. The standard approach for these calculations is time-dependent density functional theory (TDDFT) with a hybrid exchange-correlation functional like B3LYP. Because of the size of these compounds, the application of more complex quantum chemistry approaches can be challenging. In this work, we used many-body perturbation theory approaches, in particular the GW approximation with the Bethe-Salpeter equation (BSE) implemented in Gaussian basis sets, to calculate the quasiparticle properties and the absorption spectra of six cyclometalated Ir(III) complexes, going beyond TDDFT. In the presented results, we compared standard TDDFT simulations with BSE calculations performed on top of perturbative G0W0 and accounting for eigenvalue self-consistency. Moreover, in order to investigate in detail the effect of the DFT starting point, we concentrated on Ir(ppy)3 and performed GW-BSE simulations starting from different DFT exchange-correlation potentials.
中文翻译:

环金属化Ir(III)配合物的电子和光学性质的从头算多体摄动理论计算。
环金属化的Ir(III)化合物是有机发光二极管中有机发光体的首选。在实践中,过渡金属被精心设计的配体包围,可以对发射频率进行微调,并提高器件的效率。为了支持新化合物的开发,通常将实验测量值与从头算得到的吸收光谱和发射光谱进行比较。这些计算的标准方法是具有混合交换相关函数(例如B3LYP)的时变密度泛函理论(TDDFT)。由于这些化合物的大小,更复杂的量子化学方法的应用可能具有挑战性。在这项工作中,我们使用了多体摄动理论方法,尤其是用在高斯基集中实现的Bethe-Salpeter方程(BSE)进行的GW近似,以计算超出TDDFT的六个环金属化Ir(III)配合物的准粒子性质和吸收光谱。在给出的结果中,我们将标准TDDFT模拟与在摄动G0W0之上执行的BSE计算进行了比较,并考虑了特征值自洽。此外,为了详细研究DFT起点的影响,我们集中在Ir(ppy)3上,并从不同的DFT交换相关电位开始进行GW-BSE模拟。我们将标准TDDFT模拟与在摄动G0W0之上执行的BSE计算进行了比较,并考虑了特征值自洽。此外,为了详细研究DFT起点的影响,我们集中在Ir(ppy)3上,并从不同的DFT交换相关电位开始进行GW-BSE模拟。我们将标准TDDFT模拟与在摄动G0W0之上执行的BSE计算进行了比较,并考虑了特征值自洽。此外,为了详细研究DFT起点的影响,我们集中在Ir(ppy)3上,并从不同的DFT交换相关电位开始进行GW-BSE模拟。
更新日期:2020-01-07
中文翻译:
环金属化Ir(III)配合物的电子和光学性质的从头算多体摄动理论计算。
环金属化的Ir(III)化合物是有机发光二极管中有机发光体的首选。在实践中,过渡金属被精心设计的配体包围,可以对发射频率进行微调,并提高器件的效率。为了支持新化合物的开发,通常将实验测量值与从头算得到的吸收光谱和发射光谱进行比较。这些计算的标准方法是具有混合交换相关函数(例如B3LYP)的时变密度泛函理论(TDDFT)。由于这些化合物的大小,更复杂的量子化学方法的应用可能具有挑战性。在这项工作中,我们使用了多体摄动理论方法,尤其是用在高斯基集中实现的Bethe-Salpeter方程(BSE)进行的GW近似,以计算超出TDDFT的六个环金属化Ir(III)配合物的准粒子性质和吸收光谱。在给出的结果中,我们将标准TDDFT模拟与在摄动G0W0之上执行的BSE计算进行了比较,并考虑了特征值自洽。此外,为了详细研究DFT起点的影响,我们集中在Ir(ppy)3上,并从不同的DFT交换相关电位开始进行GW-BSE模拟。我们将标准TDDFT模拟与在摄动G0W0之上执行的BSE计算进行了比较,并考虑了特征值自洽。此外,为了详细研究DFT起点的影响,我们集中在Ir(ppy)3上,并从不同的DFT交换相关电位开始进行GW-BSE模拟。我们将标准TDDFT模拟与在摄动G0W0之上执行的BSE计算进行了比较,并考虑了特征值自洽。此外,为了详细研究DFT起点的影响,我们集中在Ir(ppy)3上,并从不同的DFT交换相关电位开始进行GW-BSE模拟。










































 京公网安备 11010802027423号
京公网安备 11010802027423号