当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Computational Predictions and Experimental Validation of Alkane Oxidative Dehydrogenation by Fe2M MOF Nodes
ACS Catalysis ( IF 11.3 ) Pub Date : 2020-01-07 , DOI: 10.1021/acscatal.9b03932 Melissa Barona , Sol Ahn , William Morris 1 , William Hoover 1 , Justin M. Notestein , Omar K. Farha 1 , Randall Q. Snurr
ACS Catalysis ( IF 11.3 ) Pub Date : 2020-01-07 , DOI: 10.1021/acscatal.9b03932 Melissa Barona , Sol Ahn , William Morris 1 , William Hoover 1 , Justin M. Notestein , Omar K. Farha 1 , Randall Q. Snurr
Affiliation
|
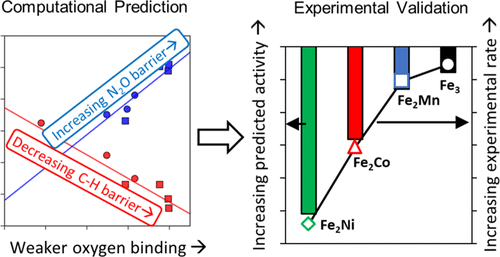
The modular structure of metal–organic frameworks (MOFs) makes them promising platforms for catalyst design and for elucidating structure/performance relationships in catalysis. In this work, we systematically varied the composition of the metal nodes (Fe2M) of the MOF PCN-250 and used density functional theory (DFT) to identify promising catalysts for light alkane C–H bond activation. Oxidative dehydrogenation (ODH) of alkanes was studied using N2O as the oxidant to understand the reactivity of the oxocentered Fe2M nodes found in PCN-250, where the Fe ions are in the +3 oxidation state and M is a metal with the oxidation state of +2. We show that the N2O activation barrier is positively correlated with the oxygen-binding energy at the metal center, and the C–H activation barrier is negatively correlated with this same quantity. For clusters containing early transition metals, oxygen binds strongly, facilitating N2O activation but hindering C–H activation. To validate the DFT predictions, we synthesized and tested PCN-250(Fe2M) with M = Mn, Fe, Co, and Ni and found that PCN-250(Fe2Mn) and PCN-250(Fe3) are more active than PCN-250(Fe2Co) and PCN-250(Fe2Ni) in agreement with the DFT predictions, demonstrating the power of DFT calculations to predict and identify promising MOF catalysts for alkane C–H bond activation in advance of experiments.
中文翻译:

Fe 2 M MOF节点对烷烃氧化脱氢的计算预测和实验验证
金属有机框架(MOF)的模块化结构使其成为催化剂设计以及阐明催化中结构/性能关系的有前途的平台。在这项工作中,我们系统地改变了MOF PCN-250的金属节点(Fe 2 M)的组成,并使用密度泛函理论(DFT)来确定有希望的轻链C-H键活化催化剂。以N 2 O为氧化剂研究了烷烃的氧化脱氢(ODH),以了解PCN-250中以氧为中心的Fe 2 M节点的反应性,其中Fe离子处于+3氧化态,M是具有+2的氧化态。我们证明N 2O激活势垒与金属中心的氧结合能成正相关,而C–H激活势垒与相同量的负相关。对于含有早期过渡金属的团簇,氧具有很强的结合力,有助于N 2 O活化,但阻碍了C–H活化。为了验证DFT预测,我们合成并测试PCN-250(铁2与M =锰,铁,Co和Ni M),并发现PCN-250(FE 2 Mn)和PCN-250(铁3)更比PCN-250(Fe 2 Co)和PCN-250(Fe 2Ni)与DFT的预测一致,证明了DFT计算的能力可以在实验之前预测和确定有前景的MOF催化剂,以促进烷烃CH键的活化。
更新日期:2020-01-07
中文翻译:
Fe 2 M MOF节点对烷烃氧化脱氢的计算预测和实验验证
金属有机框架(MOF)的模块化结构使其成为催化剂设计以及阐明催化中结构/性能关系的有前途的平台。在这项工作中,我们系统地改变了MOF PCN-250的金属节点(Fe 2 M)的组成,并使用密度泛函理论(DFT)来确定有希望的轻链C-H键活化催化剂。以N 2 O为氧化剂研究了烷烃的氧化脱氢(ODH),以了解PCN-250中以氧为中心的Fe 2 M节点的反应性,其中Fe离子处于+3氧化态,M是具有+2的氧化态。我们证明N 2O激活势垒与金属中心的氧结合能成正相关,而C–H激活势垒与相同量的负相关。对于含有早期过渡金属的团簇,氧具有很强的结合力,有助于N 2 O活化,但阻碍了C–H活化。为了验证DFT预测,我们合成并测试PCN-250(铁2与M =锰,铁,Co和Ni M),并发现PCN-250(FE 2 Mn)和PCN-250(铁3)更比PCN-250(Fe 2 Co)和PCN-250(Fe 2Ni)与DFT的预测一致,证明了DFT计算的能力可以在实验之前预测和确定有前景的MOF催化剂,以促进烷烃CH键的活化。










































 京公网安备 11010802027423号
京公网安备 11010802027423号