当前位置:
X-MOL 学术
›
Cancer Med.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Identification of prognostic genes in adrenocortical carcinoma microenvironment based on bioinformatic methods.
Cancer Medicine ( IF 2.9 ) Pub Date : 2019-12-19 , DOI: 10.1002/cam4.2774 Xiao Li 1 , Yang Gao 2 , Zicheng Xu 1 , Zheng Zhang 3 , Yuxiao Zheng 1 , Feng Qi 1, 4
Cancer Medicine ( IF 2.9 ) Pub Date : 2019-12-19 , DOI: 10.1002/cam4.2774 Xiao Li 1 , Yang Gao 2 , Zicheng Xu 1 , Zheng Zhang 3 , Yuxiao Zheng 1 , Feng Qi 1, 4
Affiliation
|
BACKGROUND
To identify prognostic genes which were associated with adrenocortical carcinoma (ACC) tumor microenvironment (TME).
METHODS AND MATERIALS
Transcriptome profiles and clinical data of ACC samples were collected from The Cancer Genome Atlas (TCGA) database. We use ESTIMATE (estimation of stromal and Immune cells in malignant tumor tissues using expression data) algorithm to calculate immune scores, stromal scores and estimate scores. Heatmap and volcano plots were applied for differential analysis. Venn plots were used for intersect genes selection. We used protein-protein interaction (PPI) networks and functional analysis to explore underlying pathways. After performing stepwise regression method and multivariate Cox analysis, we finally screened hub genes associated with ACC TME. We calculated risk scores (RS) for ACC cases based on multivariate Cox results and evaluated the prognostic value of RS shown by receiver operating characteristic curve (ROC). We investigated the association between hub genes with immune infiltrates supported by algorithm from online TIMER database.
RESULTS
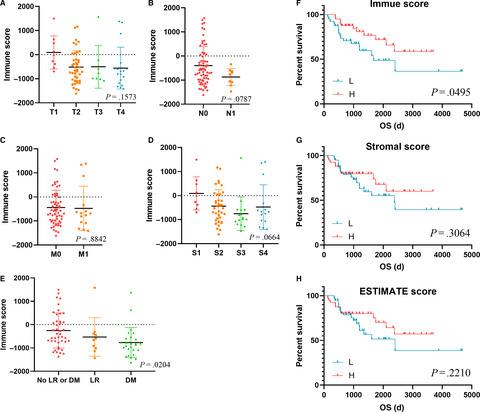
Gene expression profiles and clinical data were downloaded from TCGA. Lower immune scores were observed in disease with distant metastasis (DM) and locoregional recurrence (LR) than other cases (P = .0204). Kaplan-Meier analysis revealed that lower immune scores were significantly associated with poor overall survival (OS) (P = .0495). We screened 1649 differentially expressed genes (DEGs) and 1521 DEGs based on immune scores and stromal scores, respectively. Venn plots helped us find 1122 intersect genes. After analysing by cytoHubba from Cytoscape software, 18 hub genes were found. We calculated RS and ROC showed significantly predictive accuracy (area under curve (AUC) = 0.887). ACC patients with higher RS had worse survival outcomes (P < .0001). Results from TIMER (tumor immune estimation resource) database revealed that HLA-DOA was significantly related with immune cells infiltration.
CONCLUSION
We screened a list of TME-related genes which predict poor survival outcomes in ACC patients from TCGA database.
中文翻译:

基于生物信息学方法鉴定肾上腺皮质癌微环境中的预后基因。
背景技术为了鉴定与肾上腺皮质癌(ACC)肿瘤微环境(TME)相关的预后基因。方法和材料从癌症基因组图谱(TCGA)数据库收集了ACC样本的转录组概况和临床数据。我们使用ESTIMATE(使用表达数据估算恶性肿瘤组织中的基质细胞和免疫细胞)算法来计算免疫评分,基质评分和估算评分。使用热图和火山图进行差异分析。维恩图用于相交基因选择。我们使用蛋白质-蛋白质相互作用(PPI)网络和功能分析来探索潜在的途径。在执行逐步回归方法和多元Cox分析之后,我们最终筛选了与ACC TME相关的中枢基因。我们基于多元Cox结果计算了ACC病例的风险评分(RS),并通过接收者工作特征曲线(ROC)评估了RS的预后价值。我们研究了在线TIMER数据库中算法支持的中枢基因与免疫浸润之间的关联。结果基因表达谱和临床数据可从TCGA下载。与其他病例相比,在远处转移(DM)和局部复发(LR)的疾病中观察到较低的免疫评分(P = .0204)。Kaplan-Meier分析显示,较低的免疫评分与较差的总体生存率(OS)显着相关(P = .0495)。我们分别基于免疫评分和基质评分筛选了1649个差异表达基因(DEG)和1521个DEG。维恩图帮助我们找到了1122个相交基因。通过Cytoscape软件的cytoHubba分析后,发现了18个中枢基因。我们计算出的RS和ROC显示出显着的预测准确性(曲线下面积(AUC)= 0.887)。RS较高的ACC患者的生存结局较差(P <.0001)。TIMER(肿瘤免疫估计资源)数据库的结果显示,HLA-DOA与免疫细胞浸润显着相关。结论我们从TCGA数据库中筛选了一系列TME相关基因,这些基因可预测ACC患者的不良生存结果。TIMER(肿瘤免疫估计资源)数据库的结果显示,HLA-DOA与免疫细胞浸润显着相关。结论我们从TCGA数据库中筛选了一系列TME相关基因,这些基因可预测ACC患者的不良生存结果。TIMER(肿瘤免疫估计资源)数据库的结果显示,HLA-DOA与免疫细胞浸润显着相关。结论我们从TCGA数据库中筛选了一系列TME相关基因,这些基因可预测ACC患者的不良生存结果。
更新日期:2019-12-19
中文翻译:
基于生物信息学方法鉴定肾上腺皮质癌微环境中的预后基因。
背景技术为了鉴定与肾上腺皮质癌(ACC)肿瘤微环境(TME)相关的预后基因。方法和材料从癌症基因组图谱(TCGA)数据库收集了ACC样本的转录组概况和临床数据。我们使用ESTIMATE(使用表达数据估算恶性肿瘤组织中的基质细胞和免疫细胞)算法来计算免疫评分,基质评分和估算评分。使用热图和火山图进行差异分析。维恩图用于相交基因选择。我们使用蛋白质-蛋白质相互作用(PPI)网络和功能分析来探索潜在的途径。在执行逐步回归方法和多元Cox分析之后,我们最终筛选了与ACC TME相关的中枢基因。我们基于多元Cox结果计算了ACC病例的风险评分(RS),并通过接收者工作特征曲线(ROC)评估了RS的预后价值。我们研究了在线TIMER数据库中算法支持的中枢基因与免疫浸润之间的关联。结果基因表达谱和临床数据可从TCGA下载。与其他病例相比,在远处转移(DM)和局部复发(LR)的疾病中观察到较低的免疫评分(P = .0204)。Kaplan-Meier分析显示,较低的免疫评分与较差的总体生存率(OS)显着相关(P = .0495)。我们分别基于免疫评分和基质评分筛选了1649个差异表达基因(DEG)和1521个DEG。维恩图帮助我们找到了1122个相交基因。通过Cytoscape软件的cytoHubba分析后,发现了18个中枢基因。我们计算出的RS和ROC显示出显着的预测准确性(曲线下面积(AUC)= 0.887)。RS较高的ACC患者的生存结局较差(P <.0001)。TIMER(肿瘤免疫估计资源)数据库的结果显示,HLA-DOA与免疫细胞浸润显着相关。结论我们从TCGA数据库中筛选了一系列TME相关基因,这些基因可预测ACC患者的不良生存结果。TIMER(肿瘤免疫估计资源)数据库的结果显示,HLA-DOA与免疫细胞浸润显着相关。结论我们从TCGA数据库中筛选了一系列TME相关基因,这些基因可预测ACC患者的不良生存结果。TIMER(肿瘤免疫估计资源)数据库的结果显示,HLA-DOA与免疫细胞浸润显着相关。结论我们从TCGA数据库中筛选了一系列TME相关基因,这些基因可预测ACC患者的不良生存结果。










































 京公网安备 11010802027423号
京公网安备 11010802027423号