当前位置:
X-MOL 学术
›
Anal. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Cross-linking Mass Spectrometry Analysis of the Yeast Nucleus Reveals Extensive Protein-Protein Interactions Not Detected by Systematic Two-Hybrid or Affinity Purification-Mass Spectrometry.
Analytical Chemistry ( IF 6.7 ) Pub Date : 2020-01-09 , DOI: 10.1021/acs.analchem.9b03975 Tara K Bartolec 1 , Daniela-Lee Smith 1 , Chi Nam Ignatius Pang 1 , You Dan Xu 2 , Joshua J Hamey 1 , Marc R Wilkins 1
Analytical Chemistry ( IF 6.7 ) Pub Date : 2020-01-09 , DOI: 10.1021/acs.analchem.9b03975 Tara K Bartolec 1 , Daniela-Lee Smith 1 , Chi Nam Ignatius Pang 1 , You Dan Xu 2 , Joshua J Hamey 1 , Marc R Wilkins 1
Affiliation
|
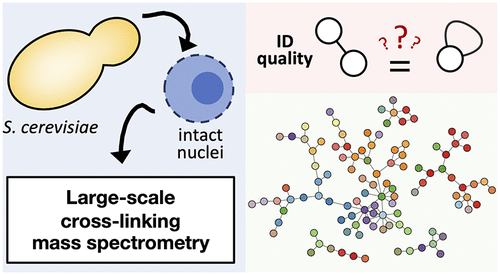
Saccharomyces cerevisiae has the most comprehensively characterized protein-protein interaction network, or interactome, of any eukaryote. This has predominantly been generated through multiple, systematic studies of protein-protein interactions by two-hybrid techniques and of affinity-purified protein complexes. A pressing question is to understand how large-scale cross-linking mass spectrometry (XL-MS) can confirm and extend this interactome. Here, intact yeast nuclei were subject to cross-linking with disuccinimidyl sulfoxide (DSSO) and analyzed using hybrid MS2-MS3 methods. XlinkX identified a total of 2,052 unique residue pair cross-links at 1% FDR. Intraprotein cross-links were found to provide extensive structural constraint data, with almost all intralinks that mapped to known structures and slightly fewer of those mapping to homology models being within 30 Å. Intralinks provided structural information for a further 366 proteins. A method for optimizing interprotein cross-link score cut-offs was developed, through use of extensive known yeast interactions. Its application led to a high confidence, yeast nuclear interactome. Strikingly, almost half of the interactions were not previously detected by two-hybrid or AP-MS techniques. Multiple lines of evidence existed for many such interactions, whether through literature or ortholog interaction data, through multiple unique interlinks between proteins, and/or through replicates. We conclude that XL-MS is a powerful means to measure interactions, that complements two-hybrid and affinity-purification techniques.
中文翻译:

酵母核的交联质谱分析揭示了广泛的蛋白质-蛋白质相互作用,而系统性的双杂交或亲和纯化-质谱检测不到。
酿酒酵母具有任何真核生物中最全面表征的蛋白质-蛋白质相互作用网络或相互作用组。这主要是通过对两种杂交技术和亲和纯化的蛋白复合物进行的蛋白-蛋白相互作用的多次系统研究而产生的。迫切需要解决的问题是了解大规模交联质谱(XL-MS)如何确认并扩展该相互作用组。在这里,完整的酵母核与二琥珀酰亚胺基亚砜(DSSO)进行交联,并使用混合MS2-MS3方法进行分析。XlinkX在1%FDR下共鉴定了2,052个独特的残基对交联。发现蛋白内交联可提供广泛的结构限制数据,具有映射到已知结构的几乎所有内部链接,而映射到同源性模型的链接几乎都在30Å之内。内部链接提供了另外366种蛋白质的结构信息。通过使用广泛已知的酵母相互作用,开发了一种优化蛋白间交联得分临界值的方法。它的应用导致了高可信度的酵母核相互作用组。令人惊讶的是,以前几乎没有通过双杂交或AP-MS技术检测到相互作用的一半。无论是通过文献还是直系同源物相互作用数据,通过蛋白质之间的多重独特相互作用和/或通过复制,对于许多此类相互作用都存在多种证据。我们得出的结论是,XL-MS是测量相互作用的有力手段,是对两种杂交技术和亲和纯化技术的补充。
更新日期:2020-01-10
中文翻译:
酵母核的交联质谱分析揭示了广泛的蛋白质-蛋白质相互作用,而系统性的双杂交或亲和纯化-质谱检测不到。
酿酒酵母具有任何真核生物中最全面表征的蛋白质-蛋白质相互作用网络或相互作用组。这主要是通过对两种杂交技术和亲和纯化的蛋白复合物进行的蛋白-蛋白相互作用的多次系统研究而产生的。迫切需要解决的问题是了解大规模交联质谱(XL-MS)如何确认并扩展该相互作用组。在这里,完整的酵母核与二琥珀酰亚胺基亚砜(DSSO)进行交联,并使用混合MS2-MS3方法进行分析。XlinkX在1%FDR下共鉴定了2,052个独特的残基对交联。发现蛋白内交联可提供广泛的结构限制数据,具有映射到已知结构的几乎所有内部链接,而映射到同源性模型的链接几乎都在30Å之内。内部链接提供了另外366种蛋白质的结构信息。通过使用广泛已知的酵母相互作用,开发了一种优化蛋白间交联得分临界值的方法。它的应用导致了高可信度的酵母核相互作用组。令人惊讶的是,以前几乎没有通过双杂交或AP-MS技术检测到相互作用的一半。无论是通过文献还是直系同源物相互作用数据,通过蛋白质之间的多重独特相互作用和/或通过复制,对于许多此类相互作用都存在多种证据。我们得出的结论是,XL-MS是测量相互作用的有力手段,是对两种杂交技术和亲和纯化技术的补充。










































 京公网安备 11010802027423号
京公网安备 11010802027423号