当前位置:
X-MOL 学术
›
Eur. J. Hum. Genet.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
SweHLA: the high confidence HLA typing bio-resource drawn from 1000 Swedish genomes.
European Journal of Human Genetics ( IF 3.7 ) Pub Date : 2019-12-16 , DOI: 10.1038/s41431-019-0559-2 Jessika Nordin 1 , Adam Ameur 2 , Kerstin Lindblad-Toh 1, 3 , Ulf Gyllensten 2 , Jennifer R S Meadows 1
European Journal of Human Genetics ( IF 3.7 ) Pub Date : 2019-12-16 , DOI: 10.1038/s41431-019-0559-2 Jessika Nordin 1 , Adam Ameur 2 , Kerstin Lindblad-Toh 1, 3 , Ulf Gyllensten 2 , Jennifer R S Meadows 1
Affiliation
|
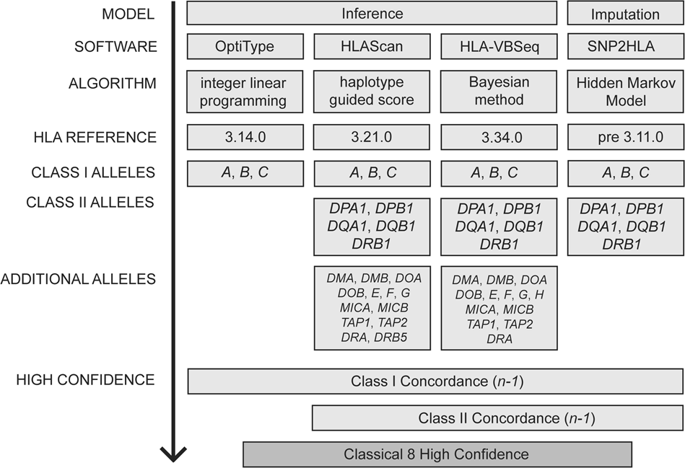
There is a need to accurately call human leukocyte antigen (HLA) genes from existing short-read sequencing data, however there is no single solution that matches the gold standard of Sanger sequenced lab typing. Here we aimed to combine results from available software programs, minimizing the biases of applied algorithm and HLA reference. The result is a robust HLA population resource for the published 1000 Swedish genomes, and a framework for future HLA interrogation. HLA 2nd-field alleles were called using four imputation and inference methods for the classical eight genes (class I: HLA-A, HLA-B, HLA-C; class II: HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1, HLA-DRB1). A high confidence population set (SweHLA) was determined using an n-1 concordance rule for class I (four software) and class II (three software) alleles. Results were compared across populations and individual programs benchmarked to SweHLA. Per gene, 875 to 988 of the 1000 samples were genotyped in SweHLA; 920 samples had at least seven loci called. While a small fraction of reference alleles were common to all software (class I = 1.9% and class II = 4.1%), this did not affect the overall call rate. Gene-level concordance was high compared to European populations (>0.83%), with COX and PGF the dominant SweHLA haplotypes. We noted that 15/18 discordant alleles (delta allele frequency >2) were previously reported as disease-associated. These differences could in part explain across-study genetic replication failures, reinforcing the need to use multiple software solutions. SweHLA demonstrates a way to use existing NGS data to generate a population resource agnostic to individual HLA software biases.
中文翻译:

SweHLA:从1000个瑞典基因组中提取的高置信度HLA分型生物资源。
有必要从现有的短读测序数据中准确调用人白细胞抗原(HLA)基因,但是没有一种解决方案符合Sanger测序实验室打字的金标准。在这里,我们旨在合并来自可用软件程序的结果,以最小化应用算法和HLA参考的偏差。结果是为已发布的1000个瑞典基因组提供了强大的HLA种群资源,并为将来的HLA审讯提供了框架。使用四种插补和推断方法对经典的八个基因调用HLA第二场等位基因(I类:HLA-A,HLA-B,HLA-C; II类:HLA-DPA1,HLA-DPB1,HLA-DQA1,HLA -DQB1,HLA-DRB1)。使用I-1类(四个软件)和II类(三个软件)等位基因的n-1一致性规则确定高置信总体集(SweHLA)。比较了不同人群和以SweHLA为基准的各个计划的结果。对于每个基因,在SweHLA中对1000个样本中的875至988个进行了基因分型;920个样本至少有7个基因座被调用。虽然一小部分参考等位基因对所有软件都是通用的(I类= 1.9%,II类= 4.1%),但这并不影响总的呼叫率。与欧洲人群相比,基因水平的一致性较高(> 0.83%),其中COX和PGF是主要的SweHLA单倍型。我们注意到先前报道15/18不等位基因(δ等位基因频率> 2)与疾病相关。这些差异可能部分解释了跨研究的基因复制失败,从而增加了使用多种软件解决方案的需求。
更新日期:2019-12-17
中文翻译:
SweHLA:从1000个瑞典基因组中提取的高置信度HLA分型生物资源。
有必要从现有的短读测序数据中准确调用人白细胞抗原(HLA)基因,但是没有一种解决方案符合Sanger测序实验室打字的金标准。在这里,我们旨在合并来自可用软件程序的结果,以最小化应用算法和HLA参考的偏差。结果是为已发布的1000个瑞典基因组提供了强大的HLA种群资源,并为将来的HLA审讯提供了框架。使用四种插补和推断方法对经典的八个基因调用HLA第二场等位基因(I类:HLA-A,HLA-B,HLA-C; II类:HLA-DPA1,HLA-DPB1,HLA-DQA1,HLA -DQB1,HLA-DRB1)。使用I-1类(四个软件)和II类(三个软件)等位基因的n-1一致性规则确定高置信总体集(SweHLA)。比较了不同人群和以SweHLA为基准的各个计划的结果。对于每个基因,在SweHLA中对1000个样本中的875至988个进行了基因分型;920个样本至少有7个基因座被调用。虽然一小部分参考等位基因对所有软件都是通用的(I类= 1.9%,II类= 4.1%),但这并不影响总的呼叫率。与欧洲人群相比,基因水平的一致性较高(> 0.83%),其中COX和PGF是主要的SweHLA单倍型。我们注意到先前报道15/18不等位基因(δ等位基因频率> 2)与疾病相关。这些差异可能部分解释了跨研究的基因复制失败,从而增加了使用多种软件解决方案的需求。










































 京公网安备 11010802027423号
京公网安备 11010802027423号