当前位置:
X-MOL 学术
›
Nat. Protoc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Genome-wide profiling of nucleosome position and chromatin accessibility in single cells using scMNase-seq.
Nature Protocols ( IF 14.8 ) Pub Date : 2019-12-13 , DOI: 10.1038/s41596-019-0243-6 Weiwu Gao 1, 2, 3, 4 , Binbin Lai 2 , Bing Ni 1, 4 , Keji Zhao 2
Nature Protocols ( IF 14.8 ) Pub Date : 2019-12-13 , DOI: 10.1038/s41596-019-0243-6 Weiwu Gao 1, 2, 3, 4 , Binbin Lai 2 , Bing Ni 1, 4 , Keji Zhao 2
Affiliation
|
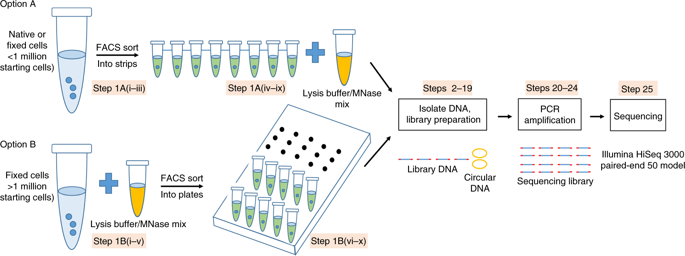
Nucleosome organization is important for chromatin compaction and accessibility. Profiling nucleosome positioning genome-wide in single cells provides critical information to understand the cell-to-cell heterogeneity of chromatin states within a cell population. This protocol describes single-cell micrococcal nuclease sequencing (scMNase-seq), a method for detecting genome-wide nucleosome positioning and chromatin accessibility simultaneously from a small number of cells or single cells. To generate scMNase-seq libraries, single cells are isolated by FACS sorting, lysed and digested by MNase. DNA is purified, end-repaired and ligated to Y-shaped adaptors. Following PCR amplification with indexing primers, the subnucleosome-sized (fragments with a length of ≤80 bp) and mononucleosome-sized (fragments with a length between 140 and 180 bp) DNA fragments are recovered and sequenced on Illumina HiSeq platforms. On average, 0.5-1 million unique mapped reads are obtained for each single cell. The mononucleosome-sized DNA fragments precisely define genome-wide nucleosome positions in single cells, while the subnucleosome-sized DNA fragments provide information on chromatin accessibility. Library preparation of scMNase-seq takes only 2 d, requires only standard molecular biology techniques and does not require sophisticated laboratory equipment. Processing of high-throughput sequencing data requires basic bioinformatics skills and uses publicly available bioinformatics software.
中文翻译:

使用scMNase-seq在单细胞中对核小体位置和染色质可及性进行全基因组分析。
核小体的组织对于染色质的紧实度和可及性很重要。在单个细胞中全基因组范围内对核小体进行定位分析可提供重要信息,以了解细胞群体内染色质状态在细胞间的异质性。该协议描述了单细胞微球菌核酸酶测序(scMNase-seq),这是一种同时从少量细胞或单个细胞中检测全基因组核小体定位和染色质可及性的方法。为了产生scMNase-seq文库,通过FACS分选分离单细胞,通过MNase裂解和消化。DNA被纯化,末端修复并与Y形衔接子连接。在使用索引引物进行PCR扩增后,回收亚核小体大小(长度≤80bp的片段)和单核小体大小(长度在140至180 bp之间的片段)的DNA片段,并在Illumina HiSeq平台上测序。平均而言,每个单个单元获得0.5-1百万个唯一的映射读段。单核小体大小的DNA片段可精确定义单个细胞中全基因组范围的核小体位置,而亚核小体大小的DNA片段可提供有关染色质可及性的信息。scMNase-seq的文库制备仅需2 d,仅需要标准分子生物学技术,而无需复杂的实验室设备。高通量测序数据的处理需要基本的生物信息学技能,并使用可公开获得的生物信息学软件。每个单个单元格获得5-1百万个唯一的映射读段。单核小体大小的DNA片段可精确定义单个细胞中全基因组范围的核小体位置,而亚核小体大小的DNA片段可提供有关染色质可及性的信息。scMNase-seq的文库制备仅需2 d,仅需要标准分子生物学技术,而无需复杂的实验室设备。高通量测序数据的处理需要基本的生物信息学技能,并使用可公开获得的生物信息学软件。每个单个单元格获得5-1百万个唯一的映射读段。单核小体大小的DNA片段可精确定义单个细胞中全基因组范围的核小体位置,而亚核小体大小的DNA片段可提供有关染色质可及性的信息。scMNase-seq的文库制备仅需2 d,仅需要标准分子生物学技术,而无需复杂的实验室设备。高通量测序数据的处理需要基本的生物信息学技能,并使用可公开获得的生物信息学软件。scMNase-seq的文库制备仅需2 d,仅需要标准分子生物学技术,而无需复杂的实验室设备。高通量测序数据的处理需要基本的生物信息学技能,并使用可公开获得的生物信息学软件。scMNase-seq的文库制备仅需2 d,仅需要标准分子生物学技术,而无需复杂的实验室设备。高通量测序数据的处理需要基本的生物信息学技能,并使用可公开获得的生物信息学软件。
更新日期:2019-12-17
中文翻译:
使用scMNase-seq在单细胞中对核小体位置和染色质可及性进行全基因组分析。
核小体的组织对于染色质的紧实度和可及性很重要。在单个细胞中全基因组范围内对核小体进行定位分析可提供重要信息,以了解细胞群体内染色质状态在细胞间的异质性。该协议描述了单细胞微球菌核酸酶测序(scMNase-seq),这是一种同时从少量细胞或单个细胞中检测全基因组核小体定位和染色质可及性的方法。为了产生scMNase-seq文库,通过FACS分选分离单细胞,通过MNase裂解和消化。DNA被纯化,末端修复并与Y形衔接子连接。在使用索引引物进行PCR扩增后,回收亚核小体大小(长度≤80bp的片段)和单核小体大小(长度在140至180 bp之间的片段)的DNA片段,并在Illumina HiSeq平台上测序。平均而言,每个单个单元获得0.5-1百万个唯一的映射读段。单核小体大小的DNA片段可精确定义单个细胞中全基因组范围的核小体位置,而亚核小体大小的DNA片段可提供有关染色质可及性的信息。scMNase-seq的文库制备仅需2 d,仅需要标准分子生物学技术,而无需复杂的实验室设备。高通量测序数据的处理需要基本的生物信息学技能,并使用可公开获得的生物信息学软件。每个单个单元格获得5-1百万个唯一的映射读段。单核小体大小的DNA片段可精确定义单个细胞中全基因组范围的核小体位置,而亚核小体大小的DNA片段可提供有关染色质可及性的信息。scMNase-seq的文库制备仅需2 d,仅需要标准分子生物学技术,而无需复杂的实验室设备。高通量测序数据的处理需要基本的生物信息学技能,并使用可公开获得的生物信息学软件。每个单个单元格获得5-1百万个唯一的映射读段。单核小体大小的DNA片段可精确定义单个细胞中全基因组范围的核小体位置,而亚核小体大小的DNA片段可提供有关染色质可及性的信息。scMNase-seq的文库制备仅需2 d,仅需要标准分子生物学技术,而无需复杂的实验室设备。高通量测序数据的处理需要基本的生物信息学技能,并使用可公开获得的生物信息学软件。scMNase-seq的文库制备仅需2 d,仅需要标准分子生物学技术,而无需复杂的实验室设备。高通量测序数据的处理需要基本的生物信息学技能,并使用可公开获得的生物信息学软件。scMNase-seq的文库制备仅需2 d,仅需要标准分子生物学技术,而无需复杂的实验室设备。高通量测序数据的处理需要基本的生物信息学技能,并使用可公开获得的生物信息学软件。


























 京公网安备 11010802027423号
京公网安备 11010802027423号