当前位置:
X-MOL 学术
›
Nat. Biotechnol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A large peptidome dataset improves HLA class I epitope prediction across most of the human population.
Nature Biotechnology ( IF 33.1 ) Pub Date : 2019-12-16 , DOI: 10.1038/s41587-019-0322-9 Siranush Sarkizova 1, 2 , Susan Klaeger 2 , Phuong M Le 3 , Letitia W Li 3 , Giacomo Oliveira 3 , Hasmik Keshishian 2 , Christina R Hartigan 2 , Wandi Zhang 3 , David A Braun 2, 3, 4, 5 , Keith L Ligon 2, 4, 6, 7 , Pavan Bachireddy 2, 3, 5 , Ioannis K Zervantonakis 8 , Jennifer M Rosenbluth 8 , Tamara Ouspenskaia 2 , Travis Law 2 , Sune Justesen 9 , Jonathan Stevens 10 , William J Lane 4, 10 , Thomas Eisenhaure 2 , Guang Lan Zhang 3, 4, 11 , Karl R Clauser 2 , Nir Hacohen 2, 3, 12 , Steven A Carr 2 , Catherine J Wu 2, 3, 4, 5 , Derin B Keskin 2, 3, 4, 5, 11
Nature Biotechnology ( IF 33.1 ) Pub Date : 2019-12-16 , DOI: 10.1038/s41587-019-0322-9 Siranush Sarkizova 1, 2 , Susan Klaeger 2 , Phuong M Le 3 , Letitia W Li 3 , Giacomo Oliveira 3 , Hasmik Keshishian 2 , Christina R Hartigan 2 , Wandi Zhang 3 , David A Braun 2, 3, 4, 5 , Keith L Ligon 2, 4, 6, 7 , Pavan Bachireddy 2, 3, 5 , Ioannis K Zervantonakis 8 , Jennifer M Rosenbluth 8 , Tamara Ouspenskaia 2 , Travis Law 2 , Sune Justesen 9 , Jonathan Stevens 10 , William J Lane 4, 10 , Thomas Eisenhaure 2 , Guang Lan Zhang 3, 4, 11 , Karl R Clauser 2 , Nir Hacohen 2, 3, 12 , Steven A Carr 2 , Catherine J Wu 2, 3, 4, 5 , Derin B Keskin 2, 3, 4, 5, 11
Affiliation
|
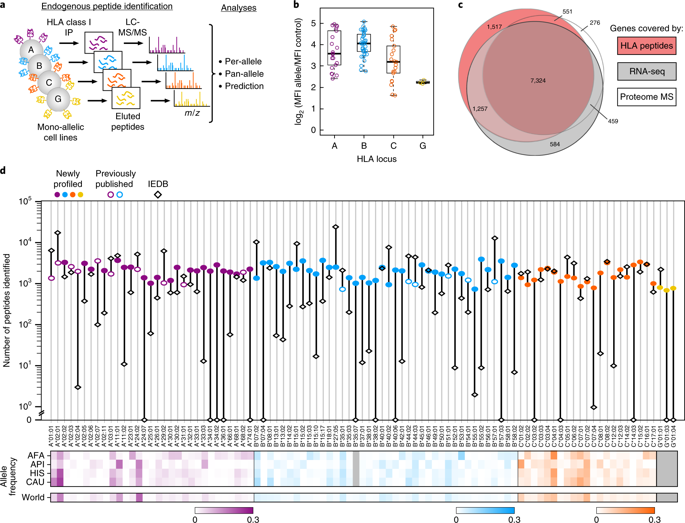
Prediction of HLA epitopes is important for the development of cancer immunotherapies and vaccines. However, current prediction algorithms have limited predictive power, in part because they were not trained on high-quality epitope datasets covering a broad range of HLA alleles. To enable prediction of endogenous HLA class I-associated peptides across a large fraction of the human population, we used mass spectrometry to profile >185,000 peptides eluted from 95 HLA-A, -B, -C and -G mono-allelic cell lines. We identified canonical peptide motifs per HLA allele, unique and shared binding submotifs across alleles and distinct motifs associated with different peptide lengths. By integrating these data with transcript abundance and peptide processing, we developed HLAthena, providing allele-and-length-specific and pan-allele-pan-length prediction models for endogenous peptide presentation. These models predicted endogenous HLA class I-associated ligands with 1.5-fold improvement in positive predictive value compared with existing tools and correctly identified >75% of HLA-bound peptides that were observed experimentally in 11 patient-derived tumor cell lines.
中文翻译:

大型肽组数据集改进了大多数人群的 HLA I 类表位预测。
HLA 表位的预测对于癌症免疫疗法和疫苗的开发非常重要。然而,当前的预测算法的预测能力有限,部分原因是它们没有接受涵盖广泛 HLA 等位基因的高质量表位数据集的训练。为了能够预测大部分人群中的内源性 HLA I 类相关肽,我们使用质谱分析了从 95 个 HLA-A、-B、-C 和 -G 单等位基因细胞中洗脱的 >185,000 个肽线。我们确定了每个 HLA 等位基因的典型肽基序、跨等位基因的独特且共享的结合子基序以及与不同肽长度相关的不同基序。通过将这些数据与转录本丰度和肽处理相结合,我们开发了 HLAthena,为内源肽呈递提供等位基因和长度特异性以及泛等位基因泛长度预测模型。与现有工具相比,这些模型预测了内源性 HLA I 类相关配体,其阳性预测值提高了 1.5 倍,并正确识别了在 11 个患者来源的肿瘤细胞系中通过实验观察到的 >75% 的 HLA 结合肽。
更新日期:2019-12-17
中文翻译:
大型肽组数据集改进了大多数人群的 HLA I 类表位预测。
HLA 表位的预测对于癌症免疫疗法和疫苗的开发非常重要。然而,当前的预测算法的预测能力有限,部分原因是它们没有接受涵盖广泛 HLA 等位基因的高质量表位数据集的训练。为了能够预测大部分人群中的内源性 HLA I 类相关肽,我们使用质谱分析了从 95 个 HLA-A、-B、-C 和 -G 单等位基因细胞中洗脱的 >185,000 个肽线。我们确定了每个 HLA 等位基因的典型肽基序、跨等位基因的独特且共享的结合子基序以及与不同肽长度相关的不同基序。通过将这些数据与转录本丰度和肽处理相结合,我们开发了 HLAthena,为内源肽呈递提供等位基因和长度特异性以及泛等位基因泛长度预测模型。与现有工具相比,这些模型预测了内源性 HLA I 类相关配体,其阳性预测值提高了 1.5 倍,并正确识别了在 11 个患者来源的肿瘤细胞系中通过实验观察到的 >75% 的 HLA 结合肽。










































 京公网安备 11010802027423号
京公网安备 11010802027423号