当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Residence Time Prediction of Type 1 and 2 Kinase Inhibitors from Unbinding Simulations.
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-01-06 , DOI: 10.1021/acs.jcim.9b00497 Abdennour Braka 1, 2 , Norbert Garnier 2 , Pascal Bonnet 1 , Samia Aci-Sèche 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2020-01-06 , DOI: 10.1021/acs.jcim.9b00497 Abdennour Braka 1, 2 , Norbert Garnier 2 , Pascal Bonnet 1 , Samia Aci-Sèche 1
Affiliation
|
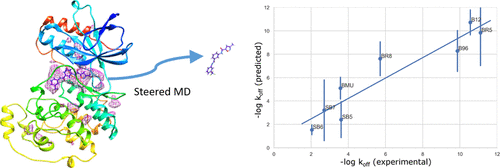
In the early stage of a drug discovery process, the selection and optimization of a ligand is mainly based on equilibrium thermodynamic constants such as KD or IC50 values, which are representatives of the affinity of the compound for its target. However, these criteria are not able to correctly evaluate the efficacy of compounds in vivo and result in many failures of compound development during phase II of clinical trials. Residence time (RT) is an important parameter associated to an in vivo drug's safety and efficacy. The determination or modulation of kinetic rates correlated to RT may be performed to identify the best drug candidates in the early stages of a drug design project. For this purpose, a number of experimental methodologies were developed but remain costly in both time and money. Herein, we developed a novel protocol based on biased molecular dynamics simulations and transition-state theory in order to predict relative ligand kinetic rates and relative RTs of a series of compounds. First, we have repeatedly simulated the unbinding process of the ligand from its binding site to the outside of the target. Next, we sample the conformational space along the determined unbinding paths to allow relevant statistical distributions of the system. The free energy profiles associated to these distributions are then computed and used to predict the kinetics parameters. The studied set was composed of eight ligands with fast, intermediate, and slow dissociation rates and binding to the active and inactive states of p38α protein kinase. The proposed method provides an excellent correlation between the predicted values and the experimentally measured kinetic rates, in addition to a detailed characterization of the kinetic paths at the atomic level.
中文翻译:

从解离模拟中预测1型和2型激酶抑制剂的停留时间。
在药物开发过程的早期阶段,配体的选择和优化主要基于平衡热力学常数(例如KD或IC50值),这些常数代表化合物对其靶标的亲和力。然而,这些标准不能正确地评估化合物在体内的功效,并导致在临床试验的第二阶段期间化合物开发的许多失败。停留时间(RT)是与体内药物的安全性和有效性相关的重要参数。可以执行与RT相关的动力学速率的确定或调节,以识别药物设计项目早期的最佳候选药物。为此目的,开发了许多实验方法,但是在时间和金钱上仍然是昂贵的。在此处,为了预测一系列化合物的相对配体动力学速率和相对RT,我们开发了一种基于偏倚分子动力学模拟和过渡态理论的新颖方案。首先,我们反复模拟了配体从其结合位点到靶标外部的解除结合过程。接下来,我们沿着确定的解绑定路径对构象空间进行采样,以允许系统的相关统计分布。然后计算与这些分布相关的自由能曲线,并将其用于预测动力学参数。该研究组由八个配体组成,这些配体具有快速,中等和缓慢的解离速率,并与p38α蛋白激酶的活性和非活性状态结合。
更新日期:2020-01-07
中文翻译:
从解离模拟中预测1型和2型激酶抑制剂的停留时间。
在药物开发过程的早期阶段,配体的选择和优化主要基于平衡热力学常数(例如KD或IC50值),这些常数代表化合物对其靶标的亲和力。然而,这些标准不能正确地评估化合物在体内的功效,并导致在临床试验的第二阶段期间化合物开发的许多失败。停留时间(RT)是与体内药物的安全性和有效性相关的重要参数。可以执行与RT相关的动力学速率的确定或调节,以识别药物设计项目早期的最佳候选药物。为此目的,开发了许多实验方法,但是在时间和金钱上仍然是昂贵的。在此处,为了预测一系列化合物的相对配体动力学速率和相对RT,我们开发了一种基于偏倚分子动力学模拟和过渡态理论的新颖方案。首先,我们反复模拟了配体从其结合位点到靶标外部的解除结合过程。接下来,我们沿着确定的解绑定路径对构象空间进行采样,以允许系统的相关统计分布。然后计算与这些分布相关的自由能曲线,并将其用于预测动力学参数。该研究组由八个配体组成,这些配体具有快速,中等和缓慢的解离速率,并与p38α蛋白激酶的活性和非活性状态结合。










































 京公网安备 11010802027423号
京公网安备 11010802027423号