当前位置:
X-MOL 学术
›
ACS Chem. Neurosci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular Dynamics Simulations Applied to Structural and Dynamical Transitions of the Huntingtin Protein: A Review.
ACS Chemical Neuroscience ( IF 4.1 ) Pub Date : 2019-12-30 , DOI: 10.1021/acschemneuro.9b00561 Sanda Nastasia Moldovean 1 , Vasile Chiş 1
ACS Chemical Neuroscience ( IF 4.1 ) Pub Date : 2019-12-30 , DOI: 10.1021/acschemneuro.9b00561 Sanda Nastasia Moldovean 1 , Vasile Chiş 1
Affiliation
|
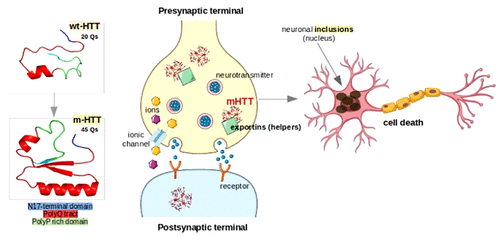
Over the recent years, Huntington's disease (HD) has become widely discussed in the scientific literature especially because at the mutant level there are several contradictions regarding the aggregation mechanism. The specific role of the physiological huntingtin protein remains unknown, due to the lack of characterization of its entire crystallographic structure, making the experimental and theoretical research even harder when taking into consideration its involvement in multiple biological functions and its high affinity for different interacting partners. Different types of models, containing fewer (not more than 35 Qs) polyglutamine residues for the WT structure and above 35 Qs for the mutants, were subjected to classical or advanced MD simulations to establish the proteins' structural stability by evaluating their conformational changes. Outside the polyQ tract, there are two other regions of interest (the N17 domain and the polyP rich domain) considered to be essential for the aggregation kinetics at the mutant level. The polymerization process is considered to be dependent on the polyQ length. As the polyQ tract's dimension increases, the structures present more β-sheet conformations. Contrarily, it is also considered that the aggregation stability is not necessarily dependent on the number of Qs, while the initial stage of the aggregation seed might play the decisive role. A general assumption regarding the polyP domain is that it might preserve the polyQ structures soluble by acting as an antagonist for β-sheet formation.
中文翻译:

分子动力学模拟应用于亨廷顿蛋白的结构和动力学转变:综述。
近年来,亨廷顿舞蹈病(HD)在科学文献中得到了广泛的讨论,特别是因为在突变体水平上,关于聚集机制存在一些矛盾。由于缺乏其整个晶体结构的表征,生理性亨廷顿蛋白的具体作用仍是未知的,考虑到其参与多种生物学功能及其对不同相互作用伙伴的高亲和力,使得其在实验和理论研究上变得更加困难。对不同类型的模型(WT结构包含较少(不超过35 Qs)聚谷氨酰胺残基,突变体包含超过35 Qs)进行经典或高级MD模拟,以建立蛋白质的 通过评估其构象变化来确定结构的稳定性。在polyQ域之外,还有两个其他感兴趣的区域(N17域和富含polyP的域)被认为对突变体水平的聚集动力学至关重要。认为聚合过程取决于polyQ长度。随着polyQ束的尺寸增加,结构呈现更多的β-折叠构象。相反,还认为聚集稳定性不一定取决于Qs的数量,而聚集种子的初始阶段可能起决定性作用。关于polyP结构域的一般假设是,它可以通过充当β-折叠形成的拮抗剂来保持polyQ结构可溶。还有其他两个感兴趣的区域(N17结构域和富含polyP的结构域)被认为对突变体水平的聚集动力学至关重要。认为聚合过程取决于polyQ长度。随着polyQ束的尺寸增加,结构呈现更多的β-折叠构象。相反,还认为聚集稳定性不一定取决于Qs的数量,而聚集种子的初始阶段可能起决定性作用。关于polyP结构域的一般假设是,它可以通过充当β-折叠形成的拮抗剂来保持polyQ结构可溶。还有其他两个感兴趣的区域(N17结构域和富含polyP的结构域)被认为对突变体水平的聚集动力学至关重要。认为聚合过程取决于polyQ长度。随着polyQ束的尺寸增加,结构呈现更多的β-折叠构象。相反,还认为聚集稳定性不一定取决于Qs的数量,而聚集种子的初始阶段可能起决定性作用。关于polyP结构域的一般假设是,它可以通过充当β-折叠形成的拮抗剂来保持polyQ结构可溶。随着polyQ束的尺寸增加,结构呈现出更多的β-折叠构象。相反,还认为聚集稳定性不一定取决于Qs的数量,而聚集种子的初始阶段可能起决定性作用。关于polyP结构域的一般假设是,它可以通过充当β-折叠形成的拮抗剂来保持polyQ结构可溶。随着polyQ束的尺寸增加,结构呈现出更多的β-折叠构象。相反,还认为聚集稳定性不一定取决于Qs的数量,而聚集种子的初始阶段可能起决定性作用。关于polyP结构域的一般假设是,它可以通过充当β-折叠形成的拮抗剂来保持polyQ结构可溶。
更新日期:2019-12-30
中文翻译:
分子动力学模拟应用于亨廷顿蛋白的结构和动力学转变:综述。
近年来,亨廷顿舞蹈病(HD)在科学文献中得到了广泛的讨论,特别是因为在突变体水平上,关于聚集机制存在一些矛盾。由于缺乏其整个晶体结构的表征,生理性亨廷顿蛋白的具体作用仍是未知的,考虑到其参与多种生物学功能及其对不同相互作用伙伴的高亲和力,使得其在实验和理论研究上变得更加困难。对不同类型的模型(WT结构包含较少(不超过35 Qs)聚谷氨酰胺残基,突变体包含超过35 Qs)进行经典或高级MD模拟,以建立蛋白质的 通过评估其构象变化来确定结构的稳定性。在polyQ域之外,还有两个其他感兴趣的区域(N17域和富含polyP的域)被认为对突变体水平的聚集动力学至关重要。认为聚合过程取决于polyQ长度。随着polyQ束的尺寸增加,结构呈现更多的β-折叠构象。相反,还认为聚集稳定性不一定取决于Qs的数量,而聚集种子的初始阶段可能起决定性作用。关于polyP结构域的一般假设是,它可以通过充当β-折叠形成的拮抗剂来保持polyQ结构可溶。还有其他两个感兴趣的区域(N17结构域和富含polyP的结构域)被认为对突变体水平的聚集动力学至关重要。认为聚合过程取决于polyQ长度。随着polyQ束的尺寸增加,结构呈现更多的β-折叠构象。相反,还认为聚集稳定性不一定取决于Qs的数量,而聚集种子的初始阶段可能起决定性作用。关于polyP结构域的一般假设是,它可以通过充当β-折叠形成的拮抗剂来保持polyQ结构可溶。还有其他两个感兴趣的区域(N17结构域和富含polyP的结构域)被认为对突变体水平的聚集动力学至关重要。认为聚合过程取决于polyQ长度。随着polyQ束的尺寸增加,结构呈现更多的β-折叠构象。相反,还认为聚集稳定性不一定取决于Qs的数量,而聚集种子的初始阶段可能起决定性作用。关于polyP结构域的一般假设是,它可以通过充当β-折叠形成的拮抗剂来保持polyQ结构可溶。随着polyQ束的尺寸增加,结构呈现出更多的β-折叠构象。相反,还认为聚集稳定性不一定取决于Qs的数量,而聚集种子的初始阶段可能起决定性作用。关于polyP结构域的一般假设是,它可以通过充当β-折叠形成的拮抗剂来保持polyQ结构可溶。随着polyQ束的尺寸增加,结构呈现出更多的β-折叠构象。相反,还认为聚集稳定性不一定取决于Qs的数量,而聚集种子的初始阶段可能起决定性作用。关于polyP结构域的一般假设是,它可以通过充当β-折叠形成的拮抗剂来保持polyQ结构可溶。










































 京公网安备 11010802027423号
京公网安备 11010802027423号