当前位置:
X-MOL 学术
›
Kidney Int.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Clinical features of genetically confirmed patients with primary hyperoxaluria identified by clinical indication versus familial screening.
Kidney International ( IF 14.8 ) Pub Date : 2019-12-13 , DOI: 10.1016/j.kint.2019.11.023 David J Sas 1 , Felicity T Enders 2 , Ramila A Mehta 2 , Xiaojing Tang 3 , Fang Zhao 4 , Barbara M Seide 4 , Dawn S Milliner 1 , John C Lieske 5
Kidney International ( IF 14.8 ) Pub Date : 2019-12-13 , DOI: 10.1016/j.kint.2019.11.023 David J Sas 1 , Felicity T Enders 2 , Ramila A Mehta 2 , Xiaojing Tang 3 , Fang Zhao 4 , Barbara M Seide 4 , Dawn S Milliner 1 , John C Lieske 5
Affiliation
|
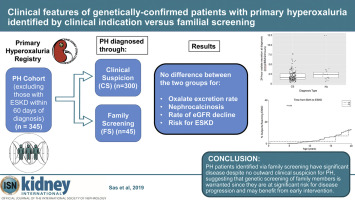
Primary hyperoxaluria is a rare monogenic disorder characterized by excessive hepatic production of oxalate leading to recurrent nephrolithiasis, nephrocalcinosis, and progressive kidney damage. Most patients with primary hyperoxaluria are diagnosed after clinical suspicion based on symptoms. Since some patients are detected by family screening following detection of an affected family member, we compared the clinical phenotype of these two groups. Patients with primary hyperoxaluria types 1, 2, and 3 enrolled in the Rare Kidney Stone Consortium Primary Hyperoxaluria Registry were retrospectively analyzed following capture of clinical and laboratory results in the Registry. Among 495 patients with primary hyperoxaluria, 47 were detected by family screening. After excluding 150 patients with end stage kidney disease at diagnosis, 300 clinical suspicion and 45 family screening individuals remained. Compared to patients with clinical suspicion, those identified by family screening had significantly fewer stones at diagnosis (mean 1.2 vs. 3.6), although initial symptoms occurred at a similar age (median age 6.1 vs. 7.6 years). Urinary oxalate did not differ between these groups. The estimated glomerular filtration rate at diagnosis and its decline over time were similar for the two groups. Altogether, five of 45 in family screening and 67 of 300 of clinical suspicion individuals developed end stage kidney disease at last follow-up. Thus, patients with primary hyperoxaluria identified through family screening have significant disease despite no outward clinical suspicion at diagnosis. Since promising novel treatments are emerging, genetic screening of family members is warranted because they are at significant risk for disease progression.
中文翻译:

通过临床适应症与家族筛查确定的经基因证实的原发性高草酸尿症患者的临床特征。
原发性高草酸尿症是一种罕见的单基因疾病,其特征是肝脏产生过多的草酸盐,导致复发性肾结石、肾钙质沉着症和进行性肾损伤。大多数原发性高草酸尿症患者是根据症状在临床怀疑后诊断的。由于在检测到受影响的家庭成员后通过家庭筛查发现了一些患者,我们比较了这两组的临床表型。对注册在罕见肾结石联盟原发性高草酸尿登记处的 1、2 和 3 型原发性高草酸尿症患者进行了回顾性分析,这些患者在登记处获取了临床和实验室结果后。495例原发性高草酸尿症患者中,47例经家庭筛查检出。在排除 150 名诊断为终末期肾病的患者后,仍有 300 名临床怀疑和 45 名家庭筛查人员。与临床怀疑的患者相比,通过家庭筛查发现的患者在诊断时结石的数量显着减少(平均 1.2 对 3.6),尽管初始症状发生的年龄相似(中位年龄 6.1 对 7.6 岁)。这些组之间的尿草酸盐没有差异。两组在诊断时估计的肾小球滤过率及其随时间的下降情况相似。总之,45 名家庭筛查中的 5 名和 300 名临床疑似患者中的 67 名在最后一次随访中发展为终末期肾病。因此,尽管在诊断时没有外在的临床怀疑,但通过家庭筛查确定的原发性高草酸尿症患者患有严重疾病。由于有前景的新疗法不断涌现,
更新日期:2019-12-13
中文翻译:
通过临床适应症与家族筛查确定的经基因证实的原发性高草酸尿症患者的临床特征。
原发性高草酸尿症是一种罕见的单基因疾病,其特征是肝脏产生过多的草酸盐,导致复发性肾结石、肾钙质沉着症和进行性肾损伤。大多数原发性高草酸尿症患者是根据症状在临床怀疑后诊断的。由于在检测到受影响的家庭成员后通过家庭筛查发现了一些患者,我们比较了这两组的临床表型。对注册在罕见肾结石联盟原发性高草酸尿登记处的 1、2 和 3 型原发性高草酸尿症患者进行了回顾性分析,这些患者在登记处获取了临床和实验室结果后。495例原发性高草酸尿症患者中,47例经家庭筛查检出。在排除 150 名诊断为终末期肾病的患者后,仍有 300 名临床怀疑和 45 名家庭筛查人员。与临床怀疑的患者相比,通过家庭筛查发现的患者在诊断时结石的数量显着减少(平均 1.2 对 3.6),尽管初始症状发生的年龄相似(中位年龄 6.1 对 7.6 岁)。这些组之间的尿草酸盐没有差异。两组在诊断时估计的肾小球滤过率及其随时间的下降情况相似。总之,45 名家庭筛查中的 5 名和 300 名临床疑似患者中的 67 名在最后一次随访中发展为终末期肾病。因此,尽管在诊断时没有外在的临床怀疑,但通过家庭筛查确定的原发性高草酸尿症患者患有严重疾病。由于有前景的新疗法不断涌现,










































 京公网安备 11010802027423号
京公网安备 11010802027423号