当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Computing Spatially Resolved Rotational Hydration Entropies from Atomistic Simulations.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2019-12-24 , DOI: 10.1021/acs.jctc.9b00926 Leonard P Heinz 1 , Helmut Grubmüller 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2019-12-24 , DOI: 10.1021/acs.jctc.9b00926 Leonard P Heinz 1 , Helmut Grubmüller 1
Affiliation
|
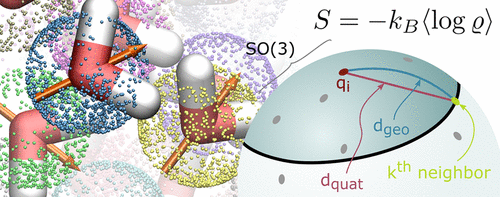
For a first-principles understanding of macromolecular processes, a quantitative understanding of the underlying free energy landscape and in particular its entropy contribution is crucial. The stability of biomolecules, such as proteins, is governed by the hydrophobic effect, which arises from competing enthalpic and entropic contributions to the free energy of the solvent shell. While the statistical mechanics of liquids, as well as molecular dynamics simulations, have provided much insight, solvation shell entropies remain notoriously difficult to calculate, especially when spatial resolution is required. Here, we present a method that allows for the computation of spatially resolved rotational solvent entropies via a nonparametric k-nearest-neighbor density estimator. We validated our method using analytic test distributions and applied it to atomistic simulations of a water box. With an accuracy of better than 9.6%, the obtained spatial resolution should shed new light on the hydrophobic effect and the thermodynamics of solvation in general.
中文翻译:

从原子模拟计算空间解析旋转水合熵。
对于大分子过程的第一性原理的理解,对潜在的自由能态尤其是其熵贡献的定量理解至关重要。生物分子(例如蛋白质)的稳定性受疏水作用的支配,该疏水作用是由于焓和熵对溶剂壳自由能的竞争贡献而产生的。尽管液体的统计力学以及分子动力学模拟提供了很多见识,但众所周知,溶剂化壳的熵仍然难以计算,尤其是在需要空间分辨率的情况下。在这里,我们提出了一种方法,该方法允许通过非参数k最近邻密度估计器来计算空间分辨的旋转溶剂熵。我们使用分析测试分布验证了我们的方法,并将其应用于水箱的原子模拟。通常,以优于9.6%的精度获得的空间分辨率应为疏水效应和溶剂化的热力学提供新的思路。
更新日期:2019-12-25
中文翻译:
从原子模拟计算空间解析旋转水合熵。
对于大分子过程的第一性原理的理解,对潜在的自由能态尤其是其熵贡献的定量理解至关重要。生物分子(例如蛋白质)的稳定性受疏水作用的支配,该疏水作用是由于焓和熵对溶剂壳自由能的竞争贡献而产生的。尽管液体的统计力学以及分子动力学模拟提供了很多见识,但众所周知,溶剂化壳的熵仍然难以计算,尤其是在需要空间分辨率的情况下。在这里,我们提出了一种方法,该方法允许通过非参数k最近邻密度估计器来计算空间分辨的旋转溶剂熵。我们使用分析测试分布验证了我们的方法,并将其应用于水箱的原子模拟。通常,以优于9.6%的精度获得的空间分辨率应为疏水效应和溶剂化的热力学提供新的思路。










































 京公网安备 11010802027423号
京公网安备 11010802027423号