Computational and Theoretical Chemistry ( IF 2.8 ) Pub Date : 2019-12-11 , DOI: 10.1016/j.comptc.2019.112669 Teepanis Chachiyo , Hathaithip Chachiyo
|
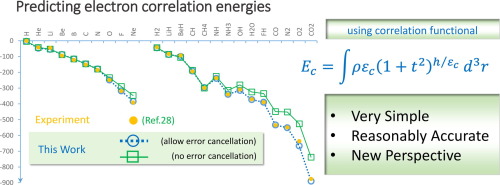
A curious behavior of electron correlation energy is explored. Namely, the correlation energy is the energy that tends to drive the system toward that of the uniform electron gas. As such, the energy assumes its maximum value when a gradient of density is zero. As the gradient increases, the energy is diminished by a gradient suppressing factor, designed to attenuate the energy from its maximum value similar to the shape of a bell curve. Based on this behavior, we constructed a very simple mathematical formula that predicted the correlation energy of atoms and molecules. Combined with our proposed exchange energy functional, we calculated the correlation, the total, the ionization, and the atomization energies of test atoms and molecules; and despite the unique simplicities, the functionals’ accuracies are in the top tier performance, competitive to the B3LYP, BLYP, PBE, TPSS, and M11.
中文翻译:
通过密度泛函理论了解电子相关能
探索了一种电子相关能量的奇怪行为。即,相关能是趋向于将系统朝着均匀电子气的方向驱动的能量。这样,当密度梯度为零时,能量取其最大值。随着梯度的增加,能量会通过梯度抑制因子而减小,该梯度抑制因子旨在使能量从其最大值(类似于钟形曲线的形状)衰减。基于此行为,我们构建了一个非常简单的数学公式,该公式可以预测原子和分子的相关能。结合我们提出的交换能量泛函,我们计算了测试原子和分子的相关性,总能量,电离能和雾化能;尽管简单明了,但功能的准确性却体现在顶级性能上,


























 京公网安备 11010802027423号
京公网安备 11010802027423号