npj Quantum Information ( IF 6.6 ) Pub Date : 2019-12-11 , DOI: 10.1038/s41534-019-0213-4 Thomas E. O’Brien , Bruno Senjean , Ramiro Sagastizabal , Xavier Bonet-Monroig , Alicja Dutkiewicz , Francesco Buda , Leonardo DiCarlo , Lucas Visscher
|
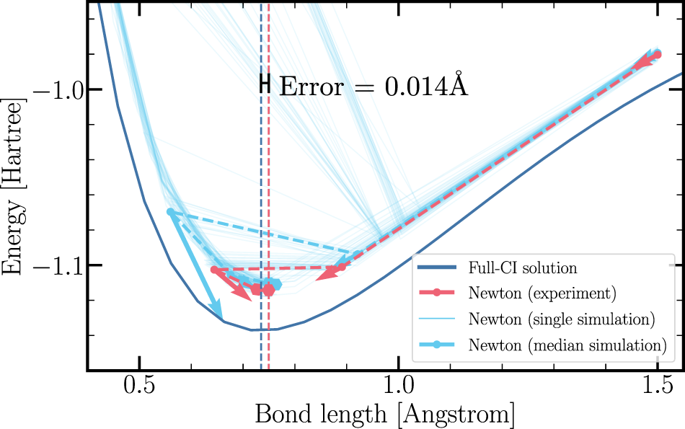
Modeling chemical reactions and complicated molecular systems has been proposed as the “killer application” of a future quantum computer. Accurate calculations of derivatives of molecular eigenenergies are essential toward this end, allowing for geometry optimization, transition state searches, predictions of the response to an applied electric or magnetic field, and molecular dynamics simulations. In this work, we survey methods to calculate energy derivatives, and present two new methods: one based on quantum phase estimation, the other on a low-order response approximation. We calculate asymptotic error bounds and approximate computational scalings for the methods presented. Implementing these methods, we perform geometry optimization on an experimental quantum processor, estimating the equilibrium bond length of the dihydrogen molecule to within \(0.014\) Å of the full configuration interaction value. Within the same experiment, we estimate the polarizability of the H\({}_{2}\) molecule, finding agreement at the equilibrium bond length to within \(0.06\) a.u. (\(2 \%\) relative error).
中文翻译:
在量子计算机上计算量子化学的能量导数
化学反应和复杂分子系统的建模已被提议作为未来量子计算机的“杀手级应用”。为此目的,精确计算分子本征能的导数是必不可少的,从而可以进行几何优化,过渡态搜索,对施加的电场或磁场的响应的预测以及分子动力学模拟。在这项工作中,我们调查了计算能量导数的方法,并提出了两种新方法:一种基于量子相位估计,另一种基于低阶响应近似。我们为提出的方法计算渐近误差范围和近似计算比例。实施这些方法后,我们在实验性量子处理器上执行几何优化, 完整配置交互值的\(0.014 \) Å。在同一实验中,我们估计H \({} _ {2} \)分子的极化率,在平衡键长度处(\(0.06 \) au(\(2 \%\)相对误差)内找到一致性。。










































 京公网安备 11010802027423号
京公网安备 11010802027423号