当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Benchmarking the Performance of Time-Dependent Density Functional Theory Methods on Biochromophores.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2019-12-26 , DOI: 10.1021/acs.jctc.9b00823 Yihan Shao 1 , Ye Mei 2, 3 , Dage Sundholm 4 , Ville R I Kaila 5, 6
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2019-12-26 , DOI: 10.1021/acs.jctc.9b00823 Yihan Shao 1 , Ye Mei 2, 3 , Dage Sundholm 4 , Ville R I Kaila 5, 6
Affiliation
|
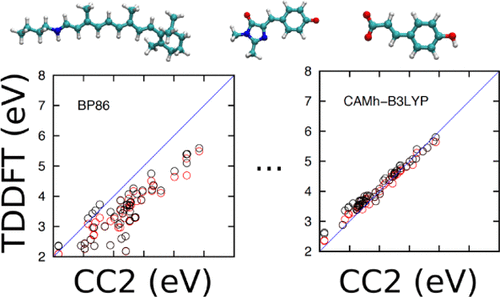
Quantum chemical calculations are important for elucidating light-capturing mechanisms in photobiological systems. The time-dependent density functional theory (TDDFT) has become a popular methodology because of its balance between accuracy and computational scaling, despite its problems in describing, for example, charge transfer states. As a step toward systematically understanding the performance of TDDFT calculations on biomolecular systems, we study here 17 commonly used density functionals, including seven long-range separated functionals, and compare the obtained results with excitation energies calculated at the approximate second order coupled-cluster theory level (CC2). The benchmarking set includes the first five singlet excited states of 11 chemical analogues of biochromophores from the green fluorescent protein, rhodopsin/bacteriorhodopsin (Rh/bR), and the photoactive yellow protein. We find that commonly used pure density functionals such as BP86, PBE, M11-L, and hybrid functionals with 20-25% of Hartree-Fock (HF) exchange (B3LYP, PBE0) have a tendency to consistently underestimate vertical excitation energies (VEEs) relative to the CC2 values, whereas hybrid density functionals with around 50% HF exchange such as BHLYP, PBE50, and M06-2X and long-range corrected functionals such as CAM-B3LYP, ωPBE, ωPBEh, ωB97X, ωB97XD, BNL, and M11 overestimate the VEEs. We observe that calculations using the CAM-B3LYP and ωPBEh functionals with 65% and 100% long-range HF exchange, respectively, lead to an overestimation of the VEEs by 0.2-0.3 eV for the benchmarking set. To reduce the systematic error, we introduce here two new empirical functionals, CAMh-B3LYP and ωhPBE0, for which we adjusted the long-range HF exchange to 50%. The introduced parameterization reduces the mean signed average (MSA) deviation to 0.07 eV and the root mean square (rms) deviation to 0.17 eV as compared to the CC2 values. In the present study, TDDFT calculations using the aug-def2-TZVP basis sets, the best performing functionals relative to CC2 are ωhPBE0 (rms = 0.17, MSA = 0.06 eV); CAMh-B3LYP (rms = 0.16, MSA = 0.07 eV); and PBE0 (rms = 0.23, MSA = -0.14 eV). For the popular range-separated CAM-B3LYP functional, we obtain an rms value of 0.31 eV and an MSA value of 0.25 eV, which can be compared with the rms and MSA values of 0.37 and -0.31 eV, respectively, as obtained at the B3LYP level.
中文翻译:

对生物发色团的时间依赖性密度泛函理论方法的性能进行基准测试。
量子化学计算对于阐明光生物系统中的光捕获机制非常重要。尽管瞬态密度泛函理论 (TDDFT) 在描述电荷转移状态等方面存在问题,但由于其在准确性和计算规模之间的平衡,它已成为一种流行的方法。作为系统地理解 TDDFT 计算在生物分子系统上的性能的一步,我们在这里研究了 17 种常用的密度泛函,包括 7 种长程分离泛函,并将获得的结果与近似二阶耦合团簇理论计算的激发能进行比较级别(CC2)。基准测试集包括来自绿色荧光蛋白、视紫红质/细菌视紫红质 (Rh/bR) 和光活性黄色蛋白的生物发色团的 11 种化学类似物的前 5 个单线态激发态。我们发现常用的纯密度泛函,例如 BP86、PBE、M11-L,以及具有 20-25% Hartree-Fock (HF) 交换的混合泛函(B3LYP、PBE0)有持续低估垂直激发能 (VEE) 的倾向。 )相对于 CC2 值,而具有大约 50% HF 交换的混合密度泛函(例如 BHLYP、PBE50 和 M06-2X)以及长程校正泛函(例如 CAM-B3LYP、ωPBE、ωPBEh、ωB97X、ωB97XD、BNL 和M11 高估了 VEE。我们观察到,使用分别具有 65% 和 100% 长程 HF 交换的 CAM-B3LYP 和 ωPBEh 泛函进行计算,导致基准测试集的 VEE 高估 0.2-0.3 eV。为了减少系统误差,我们在这里引入了两个新的经验泛函,CAMh-B3LYP 和 ωhPBE0,为此我们将长程 HF 交换调整为 50%。 与 CC2 值相比,引入的参数化将平均有符号平均值 (MSA) 偏差降低至 0.07 eV,将均方根 (rms) 偏差降低至 0.17 eV。在本研究中,使用aug-def2-TZVP基组进行TDDFT计算,相对于CC2性能最好的泛函是ωhPBE0(rms = 0.17,MSA = 0.06 eV); CAMh-B3LYP(rms = 0.16,MSA = 0.07 eV);和 PBE0(rms = 0.23,MSA = -0.14 eV)。对于流行的范围分离 CAM-B3LYP 泛函,我们获得了 0.31 eV 的 rms 值和 0.25 eV 的 MSA 值,这可以与分别在 0.37 和 -0.31 eV 的 rms 和 MSA 值进行比较。 B3LYP 级别。
更新日期:2019-12-27
中文翻译:
对生物发色团的时间依赖性密度泛函理论方法的性能进行基准测试。
量子化学计算对于阐明光生物系统中的光捕获机制非常重要。尽管瞬态密度泛函理论 (TDDFT) 在描述电荷转移状态等方面存在问题,但由于其在准确性和计算规模之间的平衡,它已成为一种流行的方法。作为系统地理解 TDDFT 计算在生物分子系统上的性能的一步,我们在这里研究了 17 种常用的密度泛函,包括 7 种长程分离泛函,并将获得的结果与近似二阶耦合团簇理论计算的激发能进行比较级别(CC2)。基准测试集包括来自绿色荧光蛋白、视紫红质/细菌视紫红质 (Rh/bR) 和光活性黄色蛋白的生物发色团的 11 种化学类似物的前 5 个单线态激发态。我们发现常用的纯密度泛函,例如 BP86、PBE、M11-L,以及具有 20-25% Hartree-Fock (HF) 交换的混合泛函(B3LYP、PBE0)有持续低估垂直激发能 (VEE) 的倾向。 )相对于 CC2 值,而具有大约 50% HF 交换的混合密度泛函(例如 BHLYP、PBE50 和 M06-2X)以及长程校正泛函(例如 CAM-B3LYP、ωPBE、ωPBEh、ωB97X、ωB97XD、BNL 和M11 高估了 VEE。我们观察到,使用分别具有 65% 和 100% 长程 HF 交换的 CAM-B3LYP 和 ωPBEh 泛函进行计算,导致基准测试集的 VEE 高估 0.2-0.3 eV。为了减少系统误差,我们在这里引入了两个新的经验泛函,CAMh-B3LYP 和 ωhPBE0,为此我们将长程 HF 交换调整为 50%。 与 CC2 值相比,引入的参数化将平均有符号平均值 (MSA) 偏差降低至 0.07 eV,将均方根 (rms) 偏差降低至 0.17 eV。在本研究中,使用aug-def2-TZVP基组进行TDDFT计算,相对于CC2性能最好的泛函是ωhPBE0(rms = 0.17,MSA = 0.06 eV); CAMh-B3LYP(rms = 0.16,MSA = 0.07 eV);和 PBE0(rms = 0.23,MSA = -0.14 eV)。对于流行的范围分离 CAM-B3LYP 泛函,我们获得了 0.31 eV 的 rms 值和 0.25 eV 的 MSA 值,这可以与分别在 0.37 和 -0.31 eV 的 rms 和 MSA 值进行比较。 B3LYP 级别。










































 京公网安备 11010802027423号
京公网安备 11010802027423号