当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Periodic Electronic Structure Calculations with the Density Matrix Embedding Theory.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2019-12-26 , DOI: 10.1021/acs.jctc.9b00939 Hung Q Pham 1 , Matthew R Hermes 1 , Laura Gagliardi 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2019-12-26 , DOI: 10.1021/acs.jctc.9b00939 Hung Q Pham 1 , Matthew R Hermes 1 , Laura Gagliardi 1
Affiliation
|
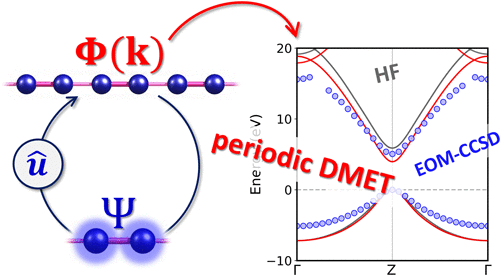
We developed a periodic version of density matrix embedding theory, DMET, with which it is possible to perform electronic structure calculations on periodic systems and compute the band structure of solid-state materials. Electron correlation can be captured by means of a local impurity model using various wave function methods, such as full configuration interaction, coupled cluster, and multiconfigurational methods. The method is able to describe not only the ground-state energy but also the quasiparticle band picture via the real space-momentum space implementation. We investigate the performance of periodic DMET in describing the ground-state energy as well as the electronic band structure for one-dimensional solids. Our results show that DMET is in good agreement with other many-body techniques at a cheaper computational cost. We anticipate that periodic DMET can be a promising first principle method for strongly correlated materials.
中文翻译:

用密度矩阵嵌入理论进行周期性电子结构计算。
我们开发了密度矩阵嵌入理论的周期性版本DMET,利用它可以在周期性系统上执行电子结构计算并计算固态材料的能带结构。可以通过使用各种波动函数方法(例如完全配置相互作用,耦合簇和多配置方法)的局部杂质模型来捕获电子相关性。该方法不仅能够通过真实的空间动量空间实现来描述基态能量,而且还能描述准粒子带图片。我们研究了周期性DMET在描述基态能量以及一维固体的电子能带结构方面的性能。我们的结果表明,DMET与其他多体技术非常吻合,但计算成本却较低。
更新日期:2019-12-27
中文翻译:
用密度矩阵嵌入理论进行周期性电子结构计算。
我们开发了密度矩阵嵌入理论的周期性版本DMET,利用它可以在周期性系统上执行电子结构计算并计算固态材料的能带结构。可以通过使用各种波动函数方法(例如完全配置相互作用,耦合簇和多配置方法)的局部杂质模型来捕获电子相关性。该方法不仅能够通过真实的空间动量空间实现来描述基态能量,而且还能描述准粒子带图片。我们研究了周期性DMET在描述基态能量以及一维固体的电子能带结构方面的性能。我们的结果表明,DMET与其他多体技术非常吻合,但计算成本却较低。










































 京公网安备 11010802027423号
京公网安备 11010802027423号