当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Rhodium-Catalyzed (5 + 2) and (5 + 1) Cycloadditions Using 1,4-Enynes as Five-Carbon Building Blocks.
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2019-12-10 , DOI: 10.1021/acs.accounts.9b00477 Stephanie A Blaszczyk 1, 2 , Daniel A Glazier 1, 2 , Weiping Tang 1, 2
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2019-12-10 , DOI: 10.1021/acs.accounts.9b00477 Stephanie A Blaszczyk 1, 2 , Daniel A Glazier 1, 2 , Weiping Tang 1, 2
Affiliation
|
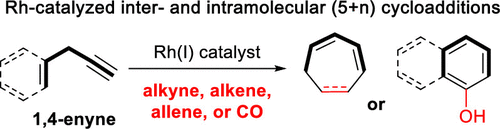
Cycloaddition reactions are a hallmark in organic synthesis because they provide an efficient way to construct highly substituted carbo- and heterocycles found in natural products and pharmaceutical agents. Most cycloadditions occur under thermal or photochemical conditions, but transition-metal complexes can promote reactions that occur beyond these circumstances. Transition-metal complexation with alkynes, alkenes, allenes, or dienes often alters the reactivity of those π-systems and facilitates access to diverse cycloaddition products. This Account describes our efforts toward the design of novel five-carbon synthons for use in rhodium-catalyzed (5 + n) cycloadditions, which include 3-acyloxy-1,4-enynes (ACEs) for (5 + 1) and (5 + 2) cycloadditions and 3-hydroxy-1,4-enynes (HYEs) for (5 + 1) cycloadditions. Furthermore, this Account includes relevant computational information, mechanistic insights, and applications of these cycloadditions in the synthesis of various highly substituted carbo- and heterocycles. The (5 + n) cycloaddition reactions presented herein share the following common mechanistic features: the 1,2-migration of an acyloxy group in propargyl esters or the ionization of a hydroxyl group in propargylic alcohols, oxidative cyclization to form a metallacycle, insertion of the one- or two-carbon component, and reductive elimination to yield the final product. In conjunction with a cationic rhodium catalyst, we used ACEs for the intramolecular (5 + 2) cycloaddition with tethered alkynes, alkenes, and allenes. In some cases, an electron-deficient phosphine ligand improved the reaction yields, especially when the ACE featured an internal alkyne. We also demonstrated that chirality could be efficiently transferred from a relatively simple starting material to a more complex bicyclic product. Products derived from ACEs with tethered alkenes and allenes contained one or more stereocenters, and high diastereoselectivity was achieved in most of these cases. For ACEs tethered to an allene, the reaction preferentially occurred at the internal alkene. We also switched the positions of the alkene and the alkyne in the 1,4-enyne of our original ACE to provide an inverted ACE variant, which produced products with complementary functionalities. After we successfully developed the Rh-catalyzed intramolecular (5 + 2) cycloaddition, we optimized conditions for the intermolecular version, which required a neutral rhodium catalyst and phosphine ligand. When a terminal alkyne was used as the two-carbon component, high regioselectivity was observed. While investigating the effect of esters on the rate of the intermolecular (5 + 2) cycloadditions, we determined that an electron-rich ester significantly accelerated the reaction. Subsequently, we demonstrated that (5 + 1) cycloadditions undergo this rate enhancement as well in the presence of an ester. Aside from ACEs, we synthesized HYEs in four steps from commercially available 2-aminobenzoic acid for use in the (5 + 1) cycloaddition. Mechanistically, HYEs were designed so that the aniline nitrogen could serve as the nucleophile and the -OH could serve as the leaving group. Using HYEs, we developed a novel method to make substituted carbazoles, dibenzofurans, and tricyclic compounds with a cyclohexadienone moiety. Although the occurrence of transition-metal-catalyzed acyloxy migrations has been known for decades, only recently has their synthetic value been realized. We hope our studies that employ readily available 1,4-enynes as the five-carbon components in (5 + n) cycloadditions can inspire the design of new two-component and multicomponent cycloadditions.
中文翻译:

使用 1,4-烯炔作为五碳结构单元进行铑催化的 (5 + 2) 和 (5 + 1) 环加成反应。
环加成反应是有机合成的一个标志,因为它们提供了一种有效的方法来构建天然产物和药剂中发现的高度取代的碳环和杂环。大多数环加成反应在热或光化学条件下发生,但过渡金属配合物可以促进在这些条件之外发生的反应。过渡金属与炔烃、烯烃、丙二烯或二烯的络合通常会改变这些π-系统的反应性,并有利于获得各种环加成产物。本报告描述了我们为设计用于铑催化 (5 + n) 环加成的新型五碳合成子所做的努力,其中包括 (5 + 1) 和 (5) 的 3-酰氧基-1,4-烯炔 (ACE) + 2) 环加成和 (5 + 1) 环加成的 3-羟基-1,4-烯炔 (HYE)。此外,本报告还包括相关的计算信息、机理见解以及这些环加成在合成各种高度取代的碳环和杂环中的应用。本文提出的 (5 + n) 环加成反应具有以下共同的机理特征:炔丙酯中酰氧基的 1,2-迁移或炔丙醇中羟基的电离、氧化环化形成金属环、插入一碳或二碳组分,并还原消除以产生最终产物。与阳离子铑催化剂结合使用,我们使用 ACE 与束缚炔烃、烯烃和丙二烯进行分子内 (5 + 2) 环加成。在某些情况下,缺电子膦配体提高了反应产率,特别是当 ACE 具有内部炔烃时。 我们还证明,手性可以有效地从相对简单的起始材料转移到更复杂的双环产物。具有束缚烯烃和丙二烯的 ACE 衍生的产物包含一个或多个立构中心,并且在大多数情况下实现了高非对映选择性。对于与丙二烯相连的 ACE,反应优先发生在内部烯烃上。我们还交换了原始 ACE 的 1,4-烯炔中烯烃和炔烃的位置,以提供反向 ACE 变体,从而产生具有互补功能的产品。在我们成功开发出铑催化的分子内 (5 + 2) 环加成反应后,我们优化了分子间环加成反应的条件,该反应需要中性铑催化剂和膦配体。当使用末端炔作为二碳组分时,观察到高区域选择性。在研究酯对分子间 (5 + 2) 环加成速率的影响时,我们确定富电子酯显着加速了反应。随后,我们证明了 (5 + 1) 环加成在酯存在的情况下也会经历这种速率增强。除了 ACE 之外,我们还通过市售 2-氨基苯甲酸分四步合成了 HYE,用于 (5 + 1) 环加成反应。从机理上讲,HYE 的设计使得苯胺氮可以充当亲核试剂,-OH 可以充当离去基团。使用 HYE,我们开发了一种新方法来制备取代咔唑、二苯并呋喃和具有环己二烯酮部分的三环化合物。尽管过渡金属催化的酰氧基迁移的发生几十年前就已为人所知,但直到最近才意识到其合成价值。 我们希望我们使用现成的 1,4-烯炔作为 (5 + n) 环加成中的五碳组分的研究能够激发新的二组分和多组分环加成的设计。
更新日期:2019-12-11
中文翻译:
使用 1,4-烯炔作为五碳结构单元进行铑催化的 (5 + 2) 和 (5 + 1) 环加成反应。
环加成反应是有机合成的一个标志,因为它们提供了一种有效的方法来构建天然产物和药剂中发现的高度取代的碳环和杂环。大多数环加成反应在热或光化学条件下发生,但过渡金属配合物可以促进在这些条件之外发生的反应。过渡金属与炔烃、烯烃、丙二烯或二烯的络合通常会改变这些π-系统的反应性,并有利于获得各种环加成产物。本报告描述了我们为设计用于铑催化 (5 + n) 环加成的新型五碳合成子所做的努力,其中包括 (5 + 1) 和 (5) 的 3-酰氧基-1,4-烯炔 (ACE) + 2) 环加成和 (5 + 1) 环加成的 3-羟基-1,4-烯炔 (HYE)。此外,本报告还包括相关的计算信息、机理见解以及这些环加成在合成各种高度取代的碳环和杂环中的应用。本文提出的 (5 + n) 环加成反应具有以下共同的机理特征:炔丙酯中酰氧基的 1,2-迁移或炔丙醇中羟基的电离、氧化环化形成金属环、插入一碳或二碳组分,并还原消除以产生最终产物。与阳离子铑催化剂结合使用,我们使用 ACE 与束缚炔烃、烯烃和丙二烯进行分子内 (5 + 2) 环加成。在某些情况下,缺电子膦配体提高了反应产率,特别是当 ACE 具有内部炔烃时。 我们还证明,手性可以有效地从相对简单的起始材料转移到更复杂的双环产物。具有束缚烯烃和丙二烯的 ACE 衍生的产物包含一个或多个立构中心,并且在大多数情况下实现了高非对映选择性。对于与丙二烯相连的 ACE,反应优先发生在内部烯烃上。我们还交换了原始 ACE 的 1,4-烯炔中烯烃和炔烃的位置,以提供反向 ACE 变体,从而产生具有互补功能的产品。在我们成功开发出铑催化的分子内 (5 + 2) 环加成反应后,我们优化了分子间环加成反应的条件,该反应需要中性铑催化剂和膦配体。当使用末端炔作为二碳组分时,观察到高区域选择性。在研究酯对分子间 (5 + 2) 环加成速率的影响时,我们确定富电子酯显着加速了反应。随后,我们证明了 (5 + 1) 环加成在酯存在的情况下也会经历这种速率增强。除了 ACE 之外,我们还通过市售 2-氨基苯甲酸分四步合成了 HYE,用于 (5 + 1) 环加成反应。从机理上讲,HYE 的设计使得苯胺氮可以充当亲核试剂,-OH 可以充当离去基团。使用 HYE,我们开发了一种新方法来制备取代咔唑、二苯并呋喃和具有环己二烯酮部分的三环化合物。尽管过渡金属催化的酰氧基迁移的发生几十年前就已为人所知,但直到最近才意识到其合成价值。 我们希望我们使用现成的 1,4-烯炔作为 (5 + n) 环加成中的五碳组分的研究能够激发新的二组分和多组分环加成的设计。










































 京公网安备 11010802027423号
京公网安备 11010802027423号