Chemical Physics ( IF 2.0 ) Pub Date : 2019-12-10 , DOI: 10.1016/j.chemphys.2019.110653 Ved Prakash Roy , Kevin J. Kubarych
|
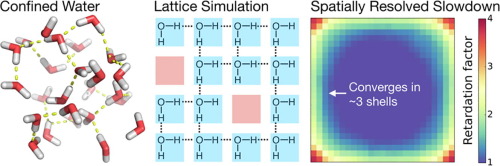
A finite, two-dimensional lattice model of liquid water captures the essential nearly four-coordinate hydrogen bonding network, while permitting a simple Metropolis Monte Carlo simulation in conditions ranging from crowded to dilute. This model examines excluded volume perturbations of hydrogen bond switching, avoiding complex topological and chemical heterogeneity of realistic interfaces. Retardation factors (relative to bulk) for switching agree with previous statistical models and atomistic molecular dynamics simulations of hydrated proteins. The model enables straightforward spatial mapping of retardation factors that are difficult to measure in atomistic simulations. The spatially-dependent retardation factors decrease exponentially from the interface. Simulating varying degrees of crowding, we do not find any cooperative, collective contributions anticipated from some recent spectroscopic observations suggesting correlated hydrogen bonding rearrangements of confined water. Longer-range cooperative interfacial influences may arise from complex chemical patterning of the surface, or to non-entropic influences such as multi-body interactions or altered hydrogen bond strengths.
中文翻译:
一个简单的格子蒙特卡洛模拟来建模界面和拥挤的水重排
液态水的有限二维晶格模型捕获了基本的接近四坐标的氢键网络,同时允许在从拥挤到稀薄的各种条件下进行简单的Metropolis Monte Carlo模拟。该模型检查了氢键转换的排除体积扰动,避免了实际界面的复杂拓扑和化学异质性。转换的延迟因子(相对于体积)与以前的统计模型和水合蛋白质的原子分子动力学模拟相吻合。该模型可以对延迟因子进行直接的空间映射,这些延迟因子在原子模拟中很难测量。取决于空间的延迟因子从界面呈指数下降。模拟不同程度的拥挤情况,我们找不到任何合作社,从最近的一些光谱观察中可以预见到集体的贡献,表明承压水的氢键重排相关。表面的复杂化学构图或非熵的影响(例如多体相互作用或氢键强度的改变)可能产生更广泛的协作界面影响。










































 京公网安备 11010802027423号
京公网安备 11010802027423号