当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Machine Learning-Guided Approach for Studying Solvation Environments.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2019-12-23 , DOI: 10.1021/acs.jctc.9b00605 Yasemin Basdogan 1 , Mitchell C Groenenboom 1 , Ethan Henderson 1 , Sandip De 2 , Susan B Rempe 3 , John A Keith 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2019-12-23 , DOI: 10.1021/acs.jctc.9b00605 Yasemin Basdogan 1 , Mitchell C Groenenboom 1 , Ethan Henderson 1 , Sandip De 2 , Susan B Rempe 3 , John A Keith 1
Affiliation
|
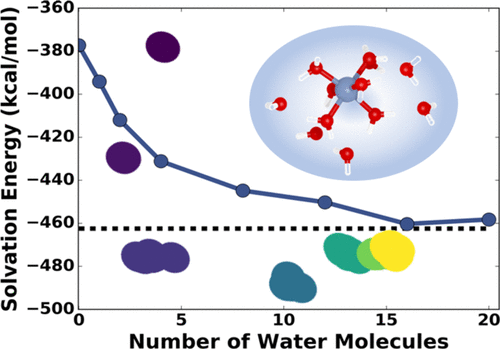
Molecular-level understanding and characterization of solvation environments are often needed across chemistry, biology, and engineering. Toward practical modeling of local solvation effects of any solute in any solvent, we report a static and all-quantum mechanics-based cluster-continuum approach for calculating single-ion solvation free energies. This approach uses a global optimization procedure to identify low-energy molecular clusters with different numbers of explicit solvent molecules and then employs the smooth overlap for atomic positions learning kernel to quantify the similarity between different low-energy solute environments. From these data, we use sketch maps, a nonlinear dimensionality reduction algorithm, to obtain a two-dimensional visual representation of the similarity between solute environments in differently sized microsolvated clusters. After testing this approach on different ions having charges 2+, 1+, 1-, and 2-, we find that the solvation environment around each ion can be seen to usually become more similar in hand with its calculated single-ion solvation free energy. Without needing either dynamics simulations or an a priori knowledge of local solvation structure of the ions, this approach can be used to calculate solvation free energies within 5% of experimental measurements for most cases, and it should be transferable for the study of other systems where dynamics simulations are not easily carried out.
中文翻译:

机器学习指导的研究溶剂化环境的方法。
在化学,生物学和工程学领域,常常需要对溶剂化环境进行分子水平的理解和表征。为了对任何溶剂中任何溶质的局部溶剂化作用进行实际建模,我们报告了一种基于静态和全量子力学的簇连续谱方法来计算单离子溶剂化自由能。该方法使用全局优化过程来识别具有不同数量的显式溶剂分子的低能分子簇,然后对原子位置学习核采用平滑重叠,以量化不同低能溶质环境之间的相似性。从这些数据中,我们使用草图,非线性降维算法,以获得二维可视化表示不同大小的微溶剂团簇中溶质环境之间相似性的信息。在对具有2 +,1 +,1-和2-电荷的不同离子测试此方法后,我们发现每个离子周围的溶剂化环境通常可以通过其计算出的单离子溶剂化自由能变得更加相似。 。无需动力学模拟或离子的局部溶剂化结构的先验知识,该方法可用于在大多数情况下计算实验测量值的5%以内的溶剂化自由能,并且应可用于研究其他系统,其中动力学仿真不容易进行。我们发现,通过计算得出的单离子溶剂化自由能,通常可以看到每个离子周围的溶剂化环境变得越来越相似。无需动力学模拟或离子的局部溶剂化结构的先验知识,该方法可用于在大多数情况下计算实验测量值的5%以内的溶剂化自由能,并且应可用于研究其他系统,其中动力学仿真不容易进行。我们发现,通过计算得出的单离子溶剂化自由能,通常可以看到每个离子周围的溶剂化环境变得越来越相似。无需动力学模拟或离子的局部溶剂化结构的先验知识,该方法可用于在大多数情况下计算实验测量值的5%以内的溶剂化自由能,并且应可用于研究其他系统,其中动力学仿真不容易进行。
更新日期:2019-12-23
中文翻译:
机器学习指导的研究溶剂化环境的方法。
在化学,生物学和工程学领域,常常需要对溶剂化环境进行分子水平的理解和表征。为了对任何溶剂中任何溶质的局部溶剂化作用进行实际建模,我们报告了一种基于静态和全量子力学的簇连续谱方法来计算单离子溶剂化自由能。该方法使用全局优化过程来识别具有不同数量的显式溶剂分子的低能分子簇,然后对原子位置学习核采用平滑重叠,以量化不同低能溶质环境之间的相似性。从这些数据中,我们使用草图,非线性降维算法,以获得二维可视化表示不同大小的微溶剂团簇中溶质环境之间相似性的信息。在对具有2 +,1 +,1-和2-电荷的不同离子测试此方法后,我们发现每个离子周围的溶剂化环境通常可以通过其计算出的单离子溶剂化自由能变得更加相似。 。无需动力学模拟或离子的局部溶剂化结构的先验知识,该方法可用于在大多数情况下计算实验测量值的5%以内的溶剂化自由能,并且应可用于研究其他系统,其中动力学仿真不容易进行。我们发现,通过计算得出的单离子溶剂化自由能,通常可以看到每个离子周围的溶剂化环境变得越来越相似。无需动力学模拟或离子的局部溶剂化结构的先验知识,该方法可用于在大多数情况下计算实验测量值的5%以内的溶剂化自由能,并且应可用于研究其他系统,其中动力学仿真不容易进行。我们发现,通过计算得出的单离子溶剂化自由能,通常可以看到每个离子周围的溶剂化环境变得越来越相似。无需动力学模拟或离子的局部溶剂化结构的先验知识,该方法可用于在大多数情况下计算实验测量值的5%以内的溶剂化自由能,并且应可用于研究其他系统,其中动力学仿真不容易进行。










































 京公网安备 11010802027423号
京公网安备 11010802027423号