当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Coordination bonding in dicopper and dichromium tetrakis(μ-acetato)-diaqua complexes: Nature, strength, length, and topology
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2019-12-05 , DOI: 10.1002/jcc.26121 Michal Malček 1 , Barbora Vénosová 1 , Ingrid Puškárová 1 , Jozef Kožíšek 1 , Marián Gall 2 , Lukáš Bučinský 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2019-12-05 , DOI: 10.1002/jcc.26121 Michal Malček 1 , Barbora Vénosová 1 , Ingrid Puškárová 1 , Jozef Kožíšek 1 , Marián Gall 2 , Lukáš Bučinský 1
Affiliation
|
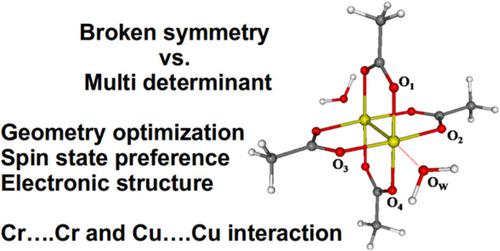
Geometry optimization, energetics, electronic structure, and topology of electron density of dicopper (I) and dichromium (II) tetrakis(μ‐acetato)‐diaqua complexes are studied focusing on the metal–metal interactions. The performance of broken symmetry (BS) single‐determinant ab initio (Hartree–Fock, Møller–Plesset perturbation theory to the second and third orders, coupled clusters singles and doubles) and density functional theory (BLYP, B3LYP, B3LYP‐D3, B2PLYP, MPW2PLYP) methods is compared to multideterminant ab initio (CASSCF, NEVPT2) methods as well as to the multipole model of charge density from a single‐crystal X‐ray diffraction experiment (Herich et al., Acta Cryst. 2018, B74, 681–692). In vacuo DFT geometry optimizations (improper axial water ligand orientation) are compared against the periodic ones. The singlet state is found to be energetically preferred. J coupling of (I) becomes underestimated for all ab initio methods used, when compared to experiment. It is concluded that the strength of the direct M─M interactions correlates closely with the J coupling magnitude at a given level of theory. The double potential well character of (II) and of the dehydrated form of (II) are considered with respect to the Cr─Cr distance. The physical effective bond order of the metal–metal interaction is small (below 0.1 e) in (I) and moderate (0.4 e) in (II). The CASSCF results overestimate the electron density of the metal–metal bond critical point by 20% and 50% in (I) and (II), respectively, when compared to the multipole model. © 2019 Wiley Periodicals, Inc.
中文翻译:

二铜和二铬四(μ-乙酰基)-diaqua 复合物中的配位键合:性质、强度、长度和拓扑结构
研究了双铜(I)和重铬(II)四(μ-乙酰)-diaqua配合物的几何优化、能量学、电子结构和电子密度拓扑,重点是金属-金属相互作用。破缺对称性 (BS) 单行列式 ab initio(Hartree-Fock、Møller-Plesset 扰动理论二阶和三阶、耦合簇单重和双重)和密度泛函理论(BLYP、B3LYP、B3LYP-D3、B2PLYP)的性能, MPW2PLYP) 方法与多行列式 ab initio (CASSCF, NEVPT2) 方法以及单晶 X 射线衍射实验中电荷密度的多极模型进行比较(Herich 等人,Acta Cryst. 2018, B74, 681 –692)。在真空中将 DFT 几何优化(不正确的轴向水配体取向)与周期性优化进行比较。发现单线态在能量上是优选的。与实验相比,(I) 的 J 耦合对于所有使用的 ab initio 方法都被低估了。得出的结论是,在给定的理论水平上,直接 M─M 相互作用的强度与 J 耦合幅度密切相关。(II) 的双势阱特性和 (II) 的脱水形式的双势阱特性是根据 Cr─Cr 距离考虑的。金属-金属相互作用的物理有效键序在(I)中很小(低于 0.1 e),在(II)中中等(0.4 e)。与多极模型相比,CASSCF 结果将 (I) 和 (II) 中金属-金属键临界点的电子密度分别高估了 20% 和 50%。© 2019 威利期刊公司。与实验相比。得出的结论是,在给定的理论水平上,直接 M─M 相互作用的强度与 J 耦合幅度密切相关。(II) 的双势阱特性和 (II) 的脱水形式的双势阱特性是根据 Cr─Cr 距离考虑的。金属-金属相互作用的物理有效键序在(I)中很小(低于 0.1 e),在(II)中中等(0.4 e)。与多极模型相比,CASSCF 结果将 (I) 和 (II) 中金属-金属键临界点的电子密度分别高估了 20% 和 50%。© 2019 威利期刊公司。与实验相比。得出的结论是,在给定的理论水平上,直接 M─M 相互作用的强度与 J 耦合幅度密切相关。(II) 的双势阱特性和 (II) 的脱水形式的双势阱特性是根据 Cr─Cr 距离考虑的。金属-金属相互作用的物理有效键序在(I)中很小(低于 0.1 e),在(II)中中等(0.4 e)。与多极模型相比,CASSCF 结果将 (I) 和 (II) 中金属-金属键临界点的电子密度分别高估了 20% 和 50%。© 2019 威利期刊公司。(II) 的双势阱特性和 (II) 的脱水形式的双势阱特性是根据 Cr─Cr 距离考虑的。金属-金属相互作用的物理有效键序在(I)中很小(低于 0.1 e),在(II)中中等(0.4 e)。与多极模型相比,CASSCF 结果将 (I) 和 (II) 中金属-金属键临界点的电子密度分别高估了 20% 和 50%。© 2019 威利期刊公司。(II) 的双势阱特性和 (II) 的脱水形式的双势阱特性是根据 Cr─Cr 距离考虑的。金属-金属相互作用的物理有效键序在(I)中很小(低于 0.1 e),在(II)中中等(0.4 e)。与多极模型相比,CASSCF 结果将 (I) 和 (II) 中金属-金属键临界点的电子密度分别高估了 20% 和 50%。© 2019 威利期刊公司。
更新日期:2019-12-05
中文翻译:
二铜和二铬四(μ-乙酰基)-diaqua 复合物中的配位键合:性质、强度、长度和拓扑结构
研究了双铜(I)和重铬(II)四(μ-乙酰)-diaqua配合物的几何优化、能量学、电子结构和电子密度拓扑,重点是金属-金属相互作用。破缺对称性 (BS) 单行列式 ab initio(Hartree-Fock、Møller-Plesset 扰动理论二阶和三阶、耦合簇单重和双重)和密度泛函理论(BLYP、B3LYP、B3LYP-D3、B2PLYP)的性能, MPW2PLYP) 方法与多行列式 ab initio (CASSCF, NEVPT2) 方法以及单晶 X 射线衍射实验中电荷密度的多极模型进行比较(Herich 等人,Acta Cryst. 2018, B74, 681 –692)。在真空中将 DFT 几何优化(不正确的轴向水配体取向)与周期性优化进行比较。发现单线态在能量上是优选的。与实验相比,(I) 的 J 耦合对于所有使用的 ab initio 方法都被低估了。得出的结论是,在给定的理论水平上,直接 M─M 相互作用的强度与 J 耦合幅度密切相关。(II) 的双势阱特性和 (II) 的脱水形式的双势阱特性是根据 Cr─Cr 距离考虑的。金属-金属相互作用的物理有效键序在(I)中很小(低于 0.1 e),在(II)中中等(0.4 e)。与多极模型相比,CASSCF 结果将 (I) 和 (II) 中金属-金属键临界点的电子密度分别高估了 20% 和 50%。© 2019 威利期刊公司。与实验相比。得出的结论是,在给定的理论水平上,直接 M─M 相互作用的强度与 J 耦合幅度密切相关。(II) 的双势阱特性和 (II) 的脱水形式的双势阱特性是根据 Cr─Cr 距离考虑的。金属-金属相互作用的物理有效键序在(I)中很小(低于 0.1 e),在(II)中中等(0.4 e)。与多极模型相比,CASSCF 结果将 (I) 和 (II) 中金属-金属键临界点的电子密度分别高估了 20% 和 50%。© 2019 威利期刊公司。与实验相比。得出的结论是,在给定的理论水平上,直接 M─M 相互作用的强度与 J 耦合幅度密切相关。(II) 的双势阱特性和 (II) 的脱水形式的双势阱特性是根据 Cr─Cr 距离考虑的。金属-金属相互作用的物理有效键序在(I)中很小(低于 0.1 e),在(II)中中等(0.4 e)。与多极模型相比,CASSCF 结果将 (I) 和 (II) 中金属-金属键临界点的电子密度分别高估了 20% 和 50%。© 2019 威利期刊公司。(II) 的双势阱特性和 (II) 的脱水形式的双势阱特性是根据 Cr─Cr 距离考虑的。金属-金属相互作用的物理有效键序在(I)中很小(低于 0.1 e),在(II)中中等(0.4 e)。与多极模型相比,CASSCF 结果将 (I) 和 (II) 中金属-金属键临界点的电子密度分别高估了 20% 和 50%。© 2019 威利期刊公司。(II) 的双势阱特性和 (II) 的脱水形式的双势阱特性是根据 Cr─Cr 距离考虑的。金属-金属相互作用的物理有效键序在(I)中很小(低于 0.1 e),在(II)中中等(0.4 e)。与多极模型相比,CASSCF 结果将 (I) 和 (II) 中金属-金属键临界点的电子密度分别高估了 20% 和 50%。© 2019 威利期刊公司。










































 京公网安备 11010802027423号
京公网安备 11010802027423号