当前位置:
X-MOL 学术
›
Acc. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Detecting Functional Dynamics in Proteins with Comparative Perturbed-Ensembles Analysis.
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2019-12-03 , DOI: 10.1021/acs.accounts.9b00485 Xin-Qiu Yao 1 , Donald Hamelberg 1
Accounts of Chemical Research ( IF 16.4 ) Pub Date : 2019-12-03 , DOI: 10.1021/acs.accounts.9b00485 Xin-Qiu Yao 1 , Donald Hamelberg 1
Affiliation
|
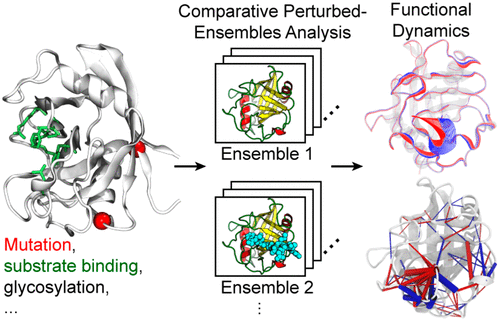
Recent advances have made all-atom molecular dynamics (MD) a powerful tool to sample the conformational energy landscape. There are still however three major challenges in the application of MD to biological systems: accuracy of force field, time scale, and the analysis of simulation trajectories. Significant progress in addressing the first two challenges has been made and extensively reviewed previously. This Account focuses on strategies of analyzing simulation data of biomolecules that also covers ways to properly design simulations and validate simulation results. In particular, we examine an approach named comparative perturbed-ensembles analysis, which we developed to efficiently detect dynamics in protein MD simulations that can be linked to biological functions. In our recent studies, we implemented this approach to understand allosteric regulations in several disease-associated human proteins. The central task of a comparative perturbed-ensembles analysis is to compare two or more conformational ensembles of a system generated by MD simulations under distinct perturbation conditions. Perturbations can be different sequence variations, ligand-binding conditions, and other physical/chemical modifications of the system. Each simulation is long enough (e.g., microsecond-long) to ensure sufficient sampling of the local substate. Then, sophisticated bioinformatic and statistical tools are applied to extract function-related information from the simulation data, including principal component analysis, residue-residue contact analysis, difference contact network analysis (dCNA) based on the graph theory, and statistical analysis of side-chain conformations. Computational findings are further validated with experimental data. By comparing distinct conformational ensembles, functional micro- to millisecond dynamics can be inferred. In contrast, such a time scale is difficult to reach in a single simulation; even when reached for a single condition of a system, it is elusive as to what dynamical motions are related to functions without, for example, comparing free and substrate-bound proteins at the minimum. We illustrate our approach with three examples. First, we discuss using the approach to identify allosteric pathways in cyclophilin A (CypA), a member of a ubiquitous class of peptidyl-prolyl cis-trans isomerase enzymes. By comparing side-chain torsion-angle distributions of CypA in wild-type and mutant forms, we identified three pathways: two are consistent with recent nuclear magnetic resonance experiments, whereas the third is a novel pathway. Second, we show how the approach enables a dynamical-evolution analysis of the human cyclophilin family. In the analysis, both conserved and divergent conformational dynamics across three cyclophilin isoforms (CypA, CypD, and CypE) were summarized. The conserved dynamics led to the discovery of allosteric networks resembling those found in CypA. A residue wise determinant underlying the unique dynamics in CypD was also detected and validated with additional mutational MD simulations. In the third example, we applied the approach to elucidate a peptide sequence-dependent allosteric mechanism in human Pin 1, a phosphorylation-dependent peptidyl-prolyl isomerase. We finally present our outlook of future directions. Especially, we envisage how the approach could help open a new avenue in drug discovery.
中文翻译:

使用比较扰动集成分析检测蛋白质中的功能动力学。
最近的进展已使全原子分子动力学(MD)成为对构象能态进行采样的强大工具。然而,将MD应用于生物系统仍然存在三个主要挑战:力场的准确性,时标和模拟轨迹的分析。在应对前两个挑战方面取得了重大进展,并且先前已进行了广泛的审查。该帐户着重于分析生物分子模拟数据的策略,该策略还涵盖了正确设计模拟和验证模拟结果的方法。特别是,我们研究了一种称为比较扰动集合分析的方法,该方法是为了有效检测可与生物学功能相关的蛋白质MD模拟中的动力学而开发的。在我们最近的研究中 我们采用这种方法来了解几种与疾病相关的人类蛋白质中的变构规律。比较扰动集合分析的中心任务是比较在不同扰动条件下通过MD模拟生成的系统的两个或多个构象集合。扰动可以是系统的不同序列变异,配体结合条件和其他物理/化学修饰。每个模拟足够长(例如,微秒长)以确保对本地子状态进行足够的采样。然后,运用先进的生物信息学和统计工具从仿真数据中提取与功能相关的信息,包括基于图论的主成分分析,残基-残基接触分析,差异接触网络分析(dCNA),和侧链构象的统计分析。计算结果可通过实验数据进一步验证。通过比较不同的构象集合,可以推断出微秒到毫秒的功能动力学。相反,在单个模拟中很难达到这样的时间范围;即使在系统的单一条件下达到要求,也无法确定哪些动态运动与功能相关,而没有(例如)比较游离蛋白和底物结合蛋白的最小值。我们通过三个示例来说明我们的方法。首先,我们讨论使用这种方法来识别亲环蛋白A(CypA)中的变构途径,亲环蛋白A(CypA)是普遍存在的肽基脯氨酰顺反异构酶的成员。通过比较野生型和突变型CypA的侧链扭转角分布,我们确定了三种途径:两项与最近的核磁共振实验一致,而第三项是一条新途径。其次,我们展示了该方法如何实现对人类亲环蛋白家族的动态进化分析。在分析中,总结了三种亲环蛋白同工型(CypA,CypD和CypE)的保守和发散构象动力学。保守的动力学导致发现了类似于CypA中发现的变构网络。还检测到了CypD中独特动力学基础的残基明智决定簇,并通过其他突变MD模拟进行了验证。在第三个示例中,我们应用了该方法来阐明人Pin 1(一种依赖于磷酸化的肽基-脯氨酰异构酶)中一种依赖肽序列的变构机制。我们终于提出了对未来方向的展望。尤其,
更新日期:2019-12-03
中文翻译:
使用比较扰动集成分析检测蛋白质中的功能动力学。
最近的进展已使全原子分子动力学(MD)成为对构象能态进行采样的强大工具。然而,将MD应用于生物系统仍然存在三个主要挑战:力场的准确性,时标和模拟轨迹的分析。在应对前两个挑战方面取得了重大进展,并且先前已进行了广泛的审查。该帐户着重于分析生物分子模拟数据的策略,该策略还涵盖了正确设计模拟和验证模拟结果的方法。特别是,我们研究了一种称为比较扰动集合分析的方法,该方法是为了有效检测可与生物学功能相关的蛋白质MD模拟中的动力学而开发的。在我们最近的研究中 我们采用这种方法来了解几种与疾病相关的人类蛋白质中的变构规律。比较扰动集合分析的中心任务是比较在不同扰动条件下通过MD模拟生成的系统的两个或多个构象集合。扰动可以是系统的不同序列变异,配体结合条件和其他物理/化学修饰。每个模拟足够长(例如,微秒长)以确保对本地子状态进行足够的采样。然后,运用先进的生物信息学和统计工具从仿真数据中提取与功能相关的信息,包括基于图论的主成分分析,残基-残基接触分析,差异接触网络分析(dCNA),和侧链构象的统计分析。计算结果可通过实验数据进一步验证。通过比较不同的构象集合,可以推断出微秒到毫秒的功能动力学。相反,在单个模拟中很难达到这样的时间范围;即使在系统的单一条件下达到要求,也无法确定哪些动态运动与功能相关,而没有(例如)比较游离蛋白和底物结合蛋白的最小值。我们通过三个示例来说明我们的方法。首先,我们讨论使用这种方法来识别亲环蛋白A(CypA)中的变构途径,亲环蛋白A(CypA)是普遍存在的肽基脯氨酰顺反异构酶的成员。通过比较野生型和突变型CypA的侧链扭转角分布,我们确定了三种途径:两项与最近的核磁共振实验一致,而第三项是一条新途径。其次,我们展示了该方法如何实现对人类亲环蛋白家族的动态进化分析。在分析中,总结了三种亲环蛋白同工型(CypA,CypD和CypE)的保守和发散构象动力学。保守的动力学导致发现了类似于CypA中发现的变构网络。还检测到了CypD中独特动力学基础的残基明智决定簇,并通过其他突变MD模拟进行了验证。在第三个示例中,我们应用了该方法来阐明人Pin 1(一种依赖于磷酸化的肽基-脯氨酰异构酶)中一种依赖肽序列的变构机制。我们终于提出了对未来方向的展望。尤其,










































 京公网安备 11010802027423号
京公网安备 11010802027423号