当前位置:
X-MOL 学术
›
Nat. Biotechnol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia.
Nature Biotechnology ( IF 33.1 ) Pub Date : 2019-12-02 , DOI: 10.1038/s41587-019-0332-7 Jeffrey M Granja 1, 2, 3 , Sandy Klemm 3 , Lisa M McGinnis 3, 4 , Arwa S Kathiria 3 , Anja Mezger 3, 5 , M Ryan Corces 1, 4 , Benjamin Parks 3, 6 , Eric Gars 4 , Michaela Liedtke 7 , Grace X Y Zheng 8 , Howard Y Chang 1, 3, 9, 10 , Ravindra Majeti 7 , William J Greenleaf 1, 3, 11, 12
Nature Biotechnology ( IF 33.1 ) Pub Date : 2019-12-02 , DOI: 10.1038/s41587-019-0332-7 Jeffrey M Granja 1, 2, 3 , Sandy Klemm 3 , Lisa M McGinnis 3, 4 , Arwa S Kathiria 3 , Anja Mezger 3, 5 , M Ryan Corces 1, 4 , Benjamin Parks 3, 6 , Eric Gars 4 , Michaela Liedtke 7 , Grace X Y Zheng 8 , Howard Y Chang 1, 3, 9, 10 , Ravindra Majeti 7 , William J Greenleaf 1, 3, 11, 12
Affiliation
|
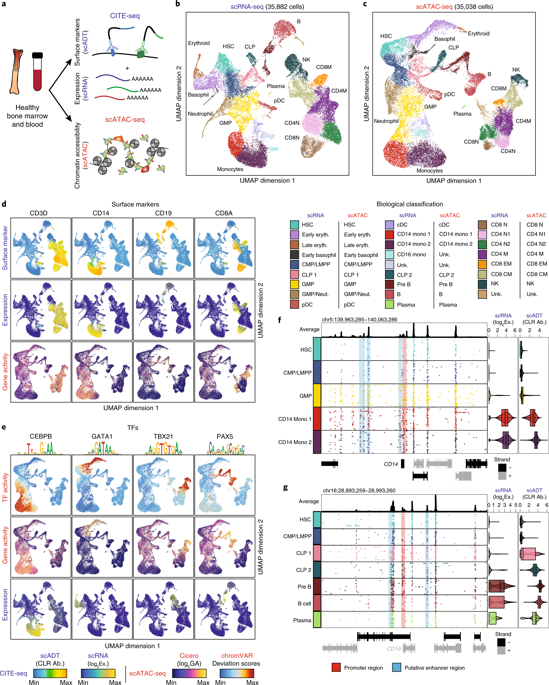
Identifying the causes of human diseases requires deconvolution of abnormal molecular phenotypes spanning DNA accessibility, gene expression and protein abundance1-3. We present a single-cell framework that integrates highly multiplexed protein quantification, transcriptome profiling and analysis of chromatin accessibility. Using this approach, we establish a normal epigenetic baseline for healthy blood development, which we then use to deconvolve aberrant molecular features within blood from patients with mixed-phenotype acute leukemia4,5. Despite widespread epigenetic heterogeneity within the patient cohort, we observe common malignant signatures across patients as well as patient-specific regulatory features that are shared across phenotypic compartments of individual patients. Integrative analysis of transcriptomic and chromatin-accessibility maps identified 91,601 putative peak-to-gene linkages and transcription factors that regulate leukemia-specific genes, such as RUNX1-linked regulatory elements proximal to the marker gene CD69. These results demonstrate how integrative, multiomic analysis of single cells within the framework of normal development can reveal both distinct and shared molecular mechanisms of disease from patient samples.
中文翻译:

单细胞多组学分析确定了混合表型急性白血病的调控程序。
确定人类疾病的原因需要对跨越 DNA 可及性、基因表达和蛋白质丰度的异常分子表型进行反卷积1-3。我们提出了一个单细胞框架,它集成了高度多路复用的蛋白质定量、转录组分析和染色质可及性分析。使用这种方法,我们为健康的血液发育建立了正常的表观遗传基线,然后我们用它来解卷积混合表型急性白血病患者血液中的异常分子特征4、5。尽管患者队列中存在广泛的表观遗传异质性,但我们观察到患者之间的共同恶性特征以及个体患者的表型区室共享的患者特异性调节特征。转录组和染色质可及性图谱的综合分析确定了 91,601 个推定的峰基因联系和调节白血病特异性基因的转录因子,例如标记基因 CD69 附近的 RUNX1 连锁调节元件。这些结果表明,在正常发育框架内对单细胞进行综合、多组学分析如何从患者样本中揭示疾病的独特和共享分子机制。
更新日期:2019-12-02
中文翻译:
单细胞多组学分析确定了混合表型急性白血病的调控程序。
确定人类疾病的原因需要对跨越 DNA 可及性、基因表达和蛋白质丰度的异常分子表型进行反卷积1-3。我们提出了一个单细胞框架,它集成了高度多路复用的蛋白质定量、转录组分析和染色质可及性分析。使用这种方法,我们为健康的血液发育建立了正常的表观遗传基线,然后我们用它来解卷积混合表型急性白血病患者血液中的异常分子特征4、5。尽管患者队列中存在广泛的表观遗传异质性,但我们观察到患者之间的共同恶性特征以及个体患者的表型区室共享的患者特异性调节特征。转录组和染色质可及性图谱的综合分析确定了 91,601 个推定的峰基因联系和调节白血病特异性基因的转录因子,例如标记基因 CD69 附近的 RUNX1 连锁调节元件。这些结果表明,在正常发育框架内对单细胞进行综合、多组学分析如何从患者样本中揭示疾病的独特和共享分子机制。










































 京公网安备 11010802027423号
京公网安备 11010802027423号