当前位置:
X-MOL 学术
›
J. Chin. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Computational insight into energetic cage derivatives based on hexahydro‐1,3,5‐trinitro‐1,3,5‐triazine
Journal of the Chinese Chemical Society ( IF 1.6 ) Pub Date : 2019-11-28 , DOI: 10.1002/jccs.201900347 Kun Wang 1 , Simin Zhu 1 , Xiaowei Wu 1 , Weihua Zhu 1
Journal of the Chinese Chemical Society ( IF 1.6 ) Pub Date : 2019-11-28 , DOI: 10.1002/jccs.201900347 Kun Wang 1 , Simin Zhu 1 , Xiaowei Wu 1 , Weihua Zhu 1
Affiliation
|
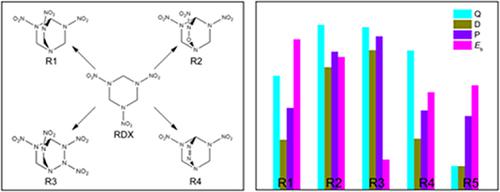
Four novel cage compounds were designed by introducing –N(NO2)CH2–, –N(NO2)O–, –N(NO2)N(NO2)–, and –N=N– linkages into the RDX (hexahydro‐1,3,5‐trinitro‐1,3,5‐triazine) skeleton. Their molecular geometry, electronic structure, heat of formation, and detonation properties were systematically studied using density functional theory (DFT). In addition, the most stable dimers of the four compounds were constructed to further investigate their stability based on intermolecular interactions. It is found that the unconventional CH⋯O interactions would be the dominant driving force when the title compounds form crystals. Compared with the traditional explosives, the compounds with higher detonation properties and lower impact sensitivity will be considered as promising candidates for high energy density compounds. Our results indicate that our innovative design strategy is extremely useful for developing novel energetic compounds.
中文翻译:

基于六氢1,3,5-三硝基-1,3,5-三嗪的高能笼型衍生物的计算洞察力
四种新型笼化合物通过引入-N(NO设计2)CH 2 - , - N(NO - 2)O - , - N(NO 2)N(NO 2)–和–N = N–连接到RDX(六氢-1,3,5-三硝基-1,3,5-三嗪)骨架中。使用密度泛函理论(DFT)系统研究了它们的分子几何结构,电子结构,形成热和爆炸特性。另外,构建了四种化合物中最稳定的二聚体,以基于分子间的相互作用进一步研究其稳定性。发现当标题化合物形成晶体时,非常规的CH = O相互作用将是主要的驱动力。与传统炸药相比,具有较高爆炸性能和较低冲击敏感性的化合物将被认为是高能量密度化合物的有前途的候选物。我们的结果表明,我们的创新设计策略对于开发新型高能化合物非常有用。
更新日期:2019-11-28
中文翻译:
基于六氢1,3,5-三硝基-1,3,5-三嗪的高能笼型衍生物的计算洞察力
四种新型笼化合物通过引入-N(NO设计2)CH 2 - , - N(NO - 2)O - , - N(NO 2)N(NO 2)–和–N = N–连接到RDX(六氢-1,3,5-三硝基-1,3,5-三嗪)骨架中。使用密度泛函理论(DFT)系统研究了它们的分子几何结构,电子结构,形成热和爆炸特性。另外,构建了四种化合物中最稳定的二聚体,以基于分子间的相互作用进一步研究其稳定性。发现当标题化合物形成晶体时,非常规的CH = O相互作用将是主要的驱动力。与传统炸药相比,具有较高爆炸性能和较低冲击敏感性的化合物将被认为是高能量密度化合物的有前途的候选物。我们的结果表明,我们的创新设计策略对于开发新型高能化合物非常有用。










































 京公网安备 11010802027423号
京公网安备 11010802027423号