European Journal of Human Genetics ( IF 3.7 ) Pub Date : 2019-11-21 , DOI: 10.1038/s41431-019-0537-8 Sergio Daga 1 , Francesco Donati 2, 3 , Katia Capitani 2, 3 , Susanna Croci 1 , Rossella Tita 4 , Annarita Giliberti 1 , Floriana Valentino 1 , Elisa Benetti 1 , Chiara Fallerini 1 , Francesca Niccheri 2 , Margherita Baldassarri 4 , Maria Antonietta Mencarelli 4 , Elisa Frullanti 1 , Simone Furini 3 , Silvestro Giovanni Conticello 2 , Alessandra Renieri 1, 4 , Anna Maria Pinto 4
|
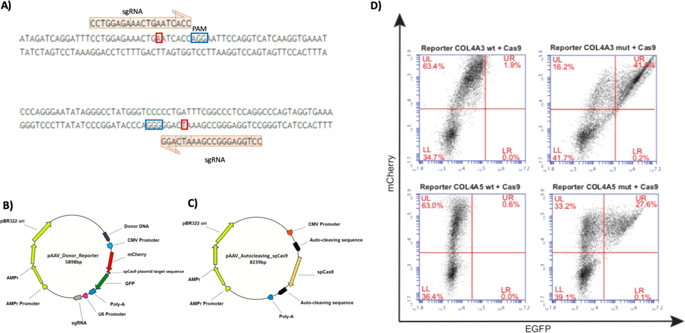
Alport syndrome (AS) is an inherited genetic disorder characterized by range of alterations from glomerular basement membrane abnormalities up to end-stage renal disease. Pathogenic variants in the collagen α3, α4, and α5 encoding genes are causative both of the autosomal dominant and of the X-linked forms of AS. Podocytes are the only renal cells that are able to produce the COL(IV)a3-a4a5 heterotrimer. We have previously demonstrated how it is possible to isolate podocyte-lineage cells from urine of patients, providing an easily accessible cellular model closer to the podocytes’ physiological conditions. Taking advantage of disease-relevant cell lines, we employed a two-plasmid approach in order to achieve a beneficial and stable variant-specific correction using CRISPR/Cas9 genome editing. One plasmid carries a Donor DNA and a reporter system mCherry/GFP to track the activity of Cas9 in cells. The other plasmid carries a self-cleaving SpCas9 and the variant-specific sgRNA. We have analyzed two stable podocyte-lineage cell lines, harboring a variant in the X-linked COL4A5 (p.(Gly624Asp)) and in the autosomal COL4A3 gene (p.(Gly856Glu)). We have achieved reversion of variants greater than 40% with undesired insertions/deletions lower than 15%. Overall, we have demonstrated a new gene therapy approach directly on patients’ cells, key players of Alport pathogenesis, and we have reverted COL4 causative variants towards the wild type state. These results, in combination with preclinical models, could open new frontiers in the management and the treatment of the disorder.
中文翻译:
治疗 Alport 综合征的新领域:足细胞谱系细胞中的 COL4A3 和 COL4A5 基因编辑。
Alport 综合征 (AS) 是一种遗传性疾病,其特征是从肾小球基底膜异常到终末期肾病的一系列改变。胶原蛋白 α3、α4 和 α5 编码基因中的致病变异是常染色体显性和 X 连锁形式 AS 的致病因素。足细胞是唯一能够产生 COL(IV)a3-a4a5 异源三聚体的肾细胞。我们之前已经证明了如何从患者的尿液中分离出足细胞系细胞,从而提供了一种更接近足细胞生理条件的易于获取的细胞模型。利用疾病相关细胞系,我们采用双质粒方法,以使用 CRISPR/Cas9 基因组编辑实现有益且稳定的变异特异性校正。一个质粒携带一个供体 DNA 和一个报告系统 mCherry/GFP 以跟踪细胞中 Cas9 的活性。另一个质粒携带自切割 SpCas9 和变体特异性 sgRNA。我们分析了两个稳定的足细胞谱系细胞系,它们在 X 连锁COL4A5 (p.(Gly624Asp)) 和常染色体COL4A3基因 (p.(Gly856Glu))。我们已经实现了大于 40% 的变体逆转,以及低于 15% 的不需要的插入/删除。总的来说,我们已经展示了一种直接针对患者细胞(Alport 发病机制的关键参与者)的新基因治疗方法,并且我们已经将COL4致病变异恢复为野生型状态。这些结果与临床前模型相结合,可以为该疾病的管理和治疗开辟新的领域。









































 京公网安备 11010802027423号
京公网安备 11010802027423号