当前位置:
X-MOL 学术
›
Int. J. Quantum Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical investigations of spin‐orbit coupling and intersystem crossing in reaction carbon dioxide activated by [Re(CO)2]+
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2019-11-19 , DOI: 10.1002/qua.26109 Juanxia Kang 1 , Yongcheng Wang 1 , Jingjing Wu 1 , Zhiming Zhu 2
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2019-11-19 , DOI: 10.1002/qua.26109 Juanxia Kang 1 , Yongcheng Wang 1 , Jingjing Wu 1 , Zhiming Zhu 2
Affiliation
|
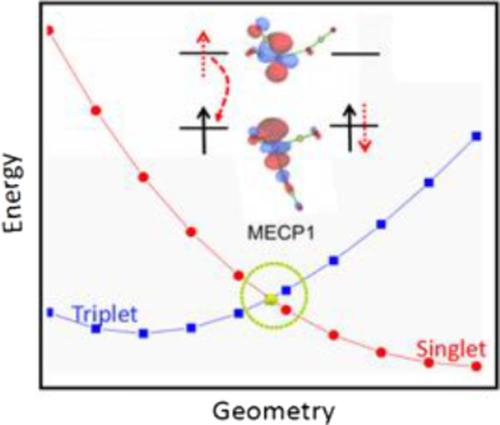
In order to further explore the detailed reaction mechanism of carbon dioxide activated by [Re(CO)2]+ complex, CCSD(T) methods was performed to determine related potential energy surface (PES). Crossing point is determined by using a partially optimized method. The result shows that larger spin‐orbital coupling (155.37 cm−1) and intersystem crossing probabilities in spin‐forbidden region causes the electron to spin flip at the minimum energy crossing point and access to the lower singlet PES. Nonadiabatic rate constant k is estimated to be quite rapid, so transition state (1TS1) is rate‐controlled steps. In addition, the electronic structure of oxygen‐atom transfer process is further analyzed by localized molecular orbital and Mayer bond order. The analysis finds that the form of main bonding orbital is the electron contribution from the p(O) in CO2 to the empty d(Re) orbital.
中文翻译:

[Re(CO)2] +活化的反应二氧化碳中自旋轨道耦合和系统间交叉的理论研究
为了进一步探讨[Re(CO)2 ] +配合物活化的二氧化碳的详细反应机理,进行了CCSD(T)法测定相关的势能面(PES)。交叉点是通过使用部分优化的方法确定的。结果表明,自旋禁区较大的自旋轨道耦合(155.37 cm -1)和系统间穿越概率导致电子在最小能量穿越点自旋翻转并进入较低的单重态PES。非绝热速率常数k估计非常快,因此过渡态(1TS1)是速率控制的步骤。此外,通过局部分子轨道和Mayer键序进一步分析了氧原子转移过程的电子结构。分析发现,主键合轨道的形式是从CO 2中的p(O)到空的d(Re)轨道的电子贡献。
更新日期:2020-01-23
中文翻译:
[Re(CO)2] +活化的反应二氧化碳中自旋轨道耦合和系统间交叉的理论研究
为了进一步探讨[Re(CO)2 ] +配合物活化的二氧化碳的详细反应机理,进行了CCSD(T)法测定相关的势能面(PES)。交叉点是通过使用部分优化的方法确定的。结果表明,自旋禁区较大的自旋轨道耦合(155.37 cm -1)和系统间穿越概率导致电子在最小能量穿越点自旋翻转并进入较低的单重态PES。非绝热速率常数k估计非常快,因此过渡态(1TS1)是速率控制的步骤。此外,通过局部分子轨道和Mayer键序进一步分析了氧原子转移过程的电子结构。分析发现,主键合轨道的形式是从CO 2中的p(O)到空的d(Re)轨道的电子贡献。










































 京公网安备 11010802027423号
京公网安备 11010802027423号