npj Schizophrenia ( IF 5.7 ) Pub Date : 2019-11-19 , DOI: 10.1038/s41537-019-0088-6 Benazir Rowe 1 , Xiangning Chen 2, 3 , Zuoheng Wang 4 , Jingchun Chen 2 , Amei Amei 1
|
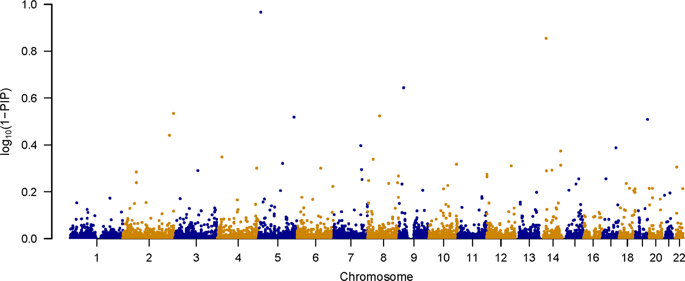
Genome-wide association studies (GWAS) have identified over 100 loci associated with schizophrenia. Most of these studies test genetic variants for association one at a time. In this study, we performed GWAS of the molecular genetics of schizophrenia (MGS) dataset with 5334 subjects using multivariate Bayesian variable selection (BVS) method Posterior Inference via Model Averaging and Subset Selection (piMASS) and compared our results with the previous univariate analysis of the MGS dataset. We showed that piMASS can improve the power of detecting schizophrenia-associated SNPs, potentially leading to new discoveries from existing data without increasing the sample size. We tested SNPs in groups to allow for local additive effects and used permutation test to determine statistical significance in order to compare our results with univariate method. The previous univariate analysis of the MGS dataset revealed no genome-wide significant loci. Using the same dataset, we identified a single region that exceeded the genome-wide significance. The result was replicated using an independent Swedish Schizophrenia Case–Control Study (SSCCS) dataset. Based on the SZGR 2.0 database we found 63 SNPs from the best performing regions that are mapped to 27 genes known to be associated with schizophrenia. Overall, we demonstrated that piMASS could discover association signals that otherwise would need a much larger sample size. Our study has important implication that reanalyzing published datasets with BVS methods like piMASS might have more power to discover new risk variants for many diseases without new sample collection, ascertainment, and genotyping.
中文翻译:
使用贝叶斯变量选择的精神分裂症全基因组关联研究的生物学和实践意义
全基因组关联研究(GWAS)已鉴定出100多个与精神分裂症有关的基因座。这些研究大多数都一次测试一种遗传变异的关联性。在这项研究中,我们使用多变量贝叶斯变量选择(BVS)方法通过模型平均和子集选择(piMASS)进行后验推断,对5334名受试者进行了精神分裂症(MGS)数据集分子遗传学的GWAS分析,并将我们的结果与之前的单变量分析进行了比较MGS数据集。我们证明了piMASS可以提高检测精神分裂症相关SNP的能力,并可能在不增加样本量的情况下从现有数据中获得新发现。我们对各组中的SNP进行了测试,以考虑局部加性效应,并使用置换检验确定统计显着性,以便将我们的结果与单变量方法进行比较。以前对MGS数据集进行的单变量分析显示,没有基因组范围内的重要基因座。使用相同的数据集,我们确定了一个超出基因组范围重要性的单个区域。使用独立的瑞典精神分裂症病例对照研究(SSCCS)数据集复制结果。基于SZGR 2.0数据库,我们从表现最佳的区域中发现了63个SNP,这些SNP被定位到已知与精神分裂症相关的27个基因上。总体而言,我们证明了piMASS可以发现关联信号,否则将需要更大的样本量。我们的研究具有重要意义,即使用piMASS等BVS方法重新分析已发布的数据集可能具有更多能力来发现许多疾病的新风险变量,而无需进行新的样本收集,确定和基因分型。










































 京公网安备 11010802027423号
京公网安备 11010802027423号