当前位置:
X-MOL 学术
›
Catal. Sci. Technol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Predicting aggregation energy for single atom bimetallic catalysts on clean and O* adsorbed surfaces through machine learning models
Catalysis Science & Technology ( IF 4.4 ) Pub Date : 2019-11-18 , DOI: 10.1039/c9cy02070e Zhuole Lu 1, 2, 3, 4 , Shwetank Yadav 1, 2, 3, 4 , Chandra Veer Singh 1, 2, 3, 4, 5
Catalysis Science & Technology ( IF 4.4 ) Pub Date : 2019-11-18 , DOI: 10.1039/c9cy02070e Zhuole Lu 1, 2, 3, 4 , Shwetank Yadav 1, 2, 3, 4 , Chandra Veer Singh 1, 2, 3, 4, 5
Affiliation
|
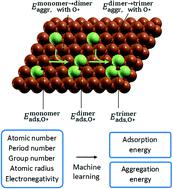
Single atom catalysts have received increased attention due to their unprecedented high metal dispersion. Recent progress in machine learning (ML) has significantly aided in the rational design of such heterogeneous catalysts, often dedicated to ML prediction of adsorption properties. A common shortfall, however, is the neglect of the thermodynamic stability of the assumed site and adsorbate-induced structural changes. Here, we developed ML models to predict the thermodynamic stability of a single-atom and related site of 38 different elements alloyed with Cu, trained with DFT calculations. To account for adsorbate-induced effects, we placed a monoatomic oxygen adsorbate in the vicinity of the dopant site. A Gaussian process regression (GPR) model performed best with a mean absolute error (MAE) of less than 0.08 eV for aggregation energy. The same performance was achieved with an even smaller training dataset using an active learning algorithm, producing a one-third time saving. Similarly, our ML prediction of O* adsorption energy gave a MAE of 0.08 eV. Furthermore, the ML model is extendable to different substrates (in addition to Cu), different adsorbates (in addition to O*), and larger cluster sizes (greater than trimer), demonstrating the potential of addressing large number of degrees of freedom and orders of magnitude time saving.
中文翻译:

通过机器学习模型预测清洁和O *吸附表面上单原子双金属催化剂的聚集能
单原子催化剂由于其前所未有的高金属分散性而受到越来越多的关注。机器学习(ML)的最新进展极大地帮助了此类多相催化剂的合理设计,这些催化剂通常专门用于ML预测吸附性能。但是,常见的不足是忽略了假定位置的热力学稳定性以及吸附剂引起的结构变化。在这里,我们开发了ML模型,以预测通过DFT计算训练的38个与Cu合金化的不同元素的单原子和相关位点的热力学稳定性。为了说明吸附物引起的影响,我们在掺杂剂位点附近放置了一个单原子氧吸附物。高斯过程回归(GPR)模型表现最佳,聚集能的平均绝对误差(MAE)小于0.08 eV。使用主动学习算法,使用更小的训练数据集也可以达到相同的性能,从而节省了三分之一的时间。同样,我们对O *吸附能的ML预测得出的MAE为0.08 eV。此外,ML模型可扩展到不同的衬底(除了Cu),不同的被吸附物(除了O *)和更大的簇尺寸(大于三聚体),证明了解决大量自由度和有序性的潜力节省大量时间。
更新日期:2019-11-18
中文翻译:
通过机器学习模型预测清洁和O *吸附表面上单原子双金属催化剂的聚集能
单原子催化剂由于其前所未有的高金属分散性而受到越来越多的关注。机器学习(ML)的最新进展极大地帮助了此类多相催化剂的合理设计,这些催化剂通常专门用于ML预测吸附性能。但是,常见的不足是忽略了假定位置的热力学稳定性以及吸附剂引起的结构变化。在这里,我们开发了ML模型,以预测通过DFT计算训练的38个与Cu合金化的不同元素的单原子和相关位点的热力学稳定性。为了说明吸附物引起的影响,我们在掺杂剂位点附近放置了一个单原子氧吸附物。高斯过程回归(GPR)模型表现最佳,聚集能的平均绝对误差(MAE)小于0.08 eV。使用主动学习算法,使用更小的训练数据集也可以达到相同的性能,从而节省了三分之一的时间。同样,我们对O *吸附能的ML预测得出的MAE为0.08 eV。此外,ML模型可扩展到不同的衬底(除了Cu),不同的被吸附物(除了O *)和更大的簇尺寸(大于三聚体),证明了解决大量自由度和有序性的潜力节省大量时间。










































 京公网安备 11010802027423号
京公网安备 11010802027423号