当前位置:
X-MOL 学术
›
Solid State Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Synthesis, crystal structure, optical absorption study, and electronic structure of Cs3FeCl5
Solid State Sciences ( IF 3.5 ) Pub Date : 2020-02-01 , DOI: 10.1016/j.solidstatesciences.2019.106064 Subhendu Jana , Gopabandhu Panigrahi , S. Narayanswamy , Mohd Ishtiyak , Manajit Das , Pinaki P. Bhattacharjee , Manish K. Niranjan , Jai Prakash
Solid State Sciences ( IF 3.5 ) Pub Date : 2020-02-01 , DOI: 10.1016/j.solidstatesciences.2019.106064 Subhendu Jana , Gopabandhu Panigrahi , S. Narayanswamy , Mohd Ishtiyak , Manajit Das , Pinaki P. Bhattacharjee , Manish K. Niranjan , Jai Prakash
|
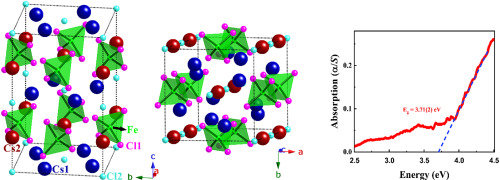
Abstract The single crystals of Cs3FeCl5 were synthesized at 973 K using the sealed tube solid-state molten flux method using CsCl as a reactive flux. The polycrystalline sample of Cs3FeCl5 was obtained by the stoichiometric reaction of CsCl and FeCl2 powders at 823 K by the sealed tube solid-state method. The crystal structure of Cs3FeCl5 was determined by single-crystal X-ray diffraction study at 298 (2) K. This ternary halide crystallizes in the body-centered tetragonal crystal system in I4/mcm space group with cell constants of a = b = 9.279 (1) A and c = 14.824 (3) A with four formula units per cell. The asymmetric unit of Cs3FeCl5 contains five crystallographically independent atomic sites: Cs1 (site symmetry: m.2 m), Cs2 (422), Fe1 ( 4 ¯ 2 m), Cl1 (..m), and Cl2 (4/m..). Each Fe atom in Cs3FeCl5 structure is bonded to four Cl1 atoms in a slightly distorted tetrahedral fashion to form isolated FeCl42− units. These FeCl42− units are separated by the Cs+ cations and infinite [CsCl] linear chains. Charge balance in this closed-shell compound can be achieved by 3 × Cs+, 1 × Fe2+, and 5 × Cl−. Bond valence sum (BVS) calculation also supports this assignment of formal oxidation states of elements in Cs3FeCl5 structure. The electronic structure calculation for Cs3FeCl5 performed within a density functional theoretical framework predicts a band gap of 3.5 eV, which is in good agreement with the experimental band gap of 3.71 (2) eV, that was estimated from the UV–vis absorption edge study of polycrystalline Cs3FeCl5.
中文翻译:

Cs3FeCl5的合成、晶体结构、光吸收研究和电子结构
摘要 Cs3FeCl5 单晶是在 973 K 下使用密封管固态熔融助熔剂法合成的,使用 CsCl 作为反应助熔剂。Cs3FeCl5 多晶样品是由 CsCl 和 FeCl2 粉末在 823 K 下通过密封管固态方法进行化学计量反应获得的。Cs3FeCl5 的晶体结构是通过 298 (2) K 下的单晶 X 射线衍射研究确定的。这种三元卤化物在 I4/mcm 空间群的体心四方晶系中结晶,晶胞常数为 a = b = 9.279 (1) A 和 c = 14.824 (3) A,每个单元格有四个公式单位。Cs3FeCl5 的不对称单元包含五个晶体学上独立的原子位点:Cs1(位点对称性:m.2 m)、Cs2 (422)、Fe1 (4¯2 m)、Cl1 (..m) 和 Cl2 (4/m)。 .) Cs3FeCl5 结构中的每个 Fe 原子以略微扭曲的四面体方式与四个 Cl1 原子键合,形成孤立的 FeCl42− 单元。这些 FeCl42- 单元被 Cs+ 阳离子和无限的 [CsCl] 线性链分开。这种闭壳化合物中的电荷平衡可以通过 3 × Cs+、1 × Fe2+ 和 5 × Cl− 实现。键价和 (BVS) 计算也支持 Cs3FeCl5 结构中元素的正式氧化态的这种分配。在密度泛函理论框架内进行的 Cs3FeCl5 的电子结构计算预测带隙为 3.5 eV,这与 3.71 (2) eV 的实验带隙非常一致,这是从 UV-vis 吸收边研究估计的多晶 Cs3FeCl5。这些 FeCl42- 单元被 Cs+ 阳离子和无限的 [CsCl] 线性链分开。这种闭壳化合物中的电荷平衡可以通过 3 × Cs+、1 × Fe2+ 和 5 × Cl− 实现。键价和 (BVS) 计算也支持 Cs3FeCl5 结构中元素的正式氧化态的这种分配。在密度泛函理论框架内进行的 Cs3FeCl5 的电子结构计算预测带隙为 3.5 eV,这与 3.71 (2) eV 的实验带隙非常一致,这是从 UV-vis 吸收边研究估计的多晶 Cs3FeCl5。这些 FeCl42- 单元被 Cs+ 阳离子和无限的 [CsCl] 线性链分开。这种闭壳化合物中的电荷平衡可以通过 3 × Cs+、1 × Fe2+ 和 5 × Cl− 实现。键价和 (BVS) 计算也支持 Cs3FeCl5 结构中元素的正式氧化态的这种分配。在密度泛函理论框架内进行的 Cs3FeCl5 的电子结构计算预测带隙为 3.5 eV,这与 3.71 (2) eV 的实验带隙非常一致,这是从 UV-vis 吸收边研究估计的多晶 Cs3FeCl5。键价和 (BVS) 计算也支持 Cs3FeCl5 结构中元素的正式氧化态的这种分配。在密度泛函理论框架内进行的 Cs3FeCl5 的电子结构计算预测带隙为 3.5 eV,这与 3.71 (2) eV 的实验带隙非常吻合,这是从 UV-vis 吸收边研究估计的多晶 Cs3FeCl5。键价和 (BVS) 计算也支持 Cs3FeCl5 结构中元素的正式氧化态的这种分配。在密度泛函理论框架内进行的 Cs3FeCl5 的电子结构计算预测带隙为 3.5 eV,这与 3.71 (2) eV 的实验带隙非常一致,这是从 UV-vis 吸收边研究估计的多晶 Cs3FeCl5。
更新日期:2020-02-01
中文翻译:
Cs3FeCl5的合成、晶体结构、光吸收研究和电子结构
摘要 Cs3FeCl5 单晶是在 973 K 下使用密封管固态熔融助熔剂法合成的,使用 CsCl 作为反应助熔剂。Cs3FeCl5 多晶样品是由 CsCl 和 FeCl2 粉末在 823 K 下通过密封管固态方法进行化学计量反应获得的。Cs3FeCl5 的晶体结构是通过 298 (2) K 下的单晶 X 射线衍射研究确定的。这种三元卤化物在 I4/mcm 空间群的体心四方晶系中结晶,晶胞常数为 a = b = 9.279 (1) A 和 c = 14.824 (3) A,每个单元格有四个公式单位。Cs3FeCl5 的不对称单元包含五个晶体学上独立的原子位点:Cs1(位点对称性:m.2 m)、Cs2 (422)、Fe1 (4¯2 m)、Cl1 (..m) 和 Cl2 (4/m)。 .) Cs3FeCl5 结构中的每个 Fe 原子以略微扭曲的四面体方式与四个 Cl1 原子键合,形成孤立的 FeCl42− 单元。这些 FeCl42- 单元被 Cs+ 阳离子和无限的 [CsCl] 线性链分开。这种闭壳化合物中的电荷平衡可以通过 3 × Cs+、1 × Fe2+ 和 5 × Cl− 实现。键价和 (BVS) 计算也支持 Cs3FeCl5 结构中元素的正式氧化态的这种分配。在密度泛函理论框架内进行的 Cs3FeCl5 的电子结构计算预测带隙为 3.5 eV,这与 3.71 (2) eV 的实验带隙非常一致,这是从 UV-vis 吸收边研究估计的多晶 Cs3FeCl5。这些 FeCl42- 单元被 Cs+ 阳离子和无限的 [CsCl] 线性链分开。这种闭壳化合物中的电荷平衡可以通过 3 × Cs+、1 × Fe2+ 和 5 × Cl− 实现。键价和 (BVS) 计算也支持 Cs3FeCl5 结构中元素的正式氧化态的这种分配。在密度泛函理论框架内进行的 Cs3FeCl5 的电子结构计算预测带隙为 3.5 eV,这与 3.71 (2) eV 的实验带隙非常一致,这是从 UV-vis 吸收边研究估计的多晶 Cs3FeCl5。这些 FeCl42- 单元被 Cs+ 阳离子和无限的 [CsCl] 线性链分开。这种闭壳化合物中的电荷平衡可以通过 3 × Cs+、1 × Fe2+ 和 5 × Cl− 实现。键价和 (BVS) 计算也支持 Cs3FeCl5 结构中元素的正式氧化态的这种分配。在密度泛函理论框架内进行的 Cs3FeCl5 的电子结构计算预测带隙为 3.5 eV,这与 3.71 (2) eV 的实验带隙非常一致,这是从 UV-vis 吸收边研究估计的多晶 Cs3FeCl5。键价和 (BVS) 计算也支持 Cs3FeCl5 结构中元素的正式氧化态的这种分配。在密度泛函理论框架内进行的 Cs3FeCl5 的电子结构计算预测带隙为 3.5 eV,这与 3.71 (2) eV 的实验带隙非常吻合,这是从 UV-vis 吸收边研究估计的多晶 Cs3FeCl5。键价和 (BVS) 计算也支持 Cs3FeCl5 结构中元素的正式氧化态的这种分配。在密度泛函理论框架内进行的 Cs3FeCl5 的电子结构计算预测带隙为 3.5 eV,这与 3.71 (2) eV 的实验带隙非常一致,这是从 UV-vis 吸收边研究估计的多晶 Cs3FeCl5。


























 京公网安备 11010802027423号
京公网安备 11010802027423号