当前位置:
X-MOL 学术
›
Vib. Spectrosc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Optimization of Synephrine and its Vibrational and Electronic Structures
Vibrational Spectroscopy ( IF 2.7 ) Pub Date : 2020-01-01 , DOI: 10.1016/j.vibspec.2019.102989 T. Yadav , V. Mukherjee
Vibrational Spectroscopy ( IF 2.7 ) Pub Date : 2020-01-01 , DOI: 10.1016/j.vibspec.2019.102989 T. Yadav , V. Mukherjee
|
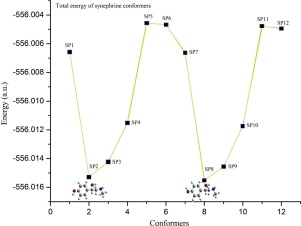
Abstract The optimization of isolated synephrine molecule and its HCl cluster was performed. The potential energy scanning with respect to some dihedral angles at ethylamine side chain was also performed which prefers twelve different low energy structures of single synephrine molecule. The effect of N..H hydrogen bond on geometry and vibrational properties of synephrine in the least energy conformer was studied. The vibrational and electronic structures of all the twelve conformers of synephrine were computed. The experimental FTIR and Raman spectra of synephrine were also recorded in the spectral region 4000–400 cm−1 and 3200-50 cm−1 respectively and correlated with the theoretical spectra of energetically most preferred structure of synephrine. The normal coordinate analysis was employed to scale the overestimated DFT frequencies and to calculate potential energy distributions of vibrtional modes of synephrine. The natural bond orbital analysis was also performed to obtain bond energies and occupancies, HOMO-LUMO and hybridization.
中文翻译:

辛弗林及其振动和电子结构的优化
摘要 对分离的辛弗林分子及其HCl簇进行了优化。还进行了关于乙胺侧链的一些二面角的势能扫描,其优选单个辛弗林分子的十二个不同低能结构。研究了 N..H 氢键对辛弗林在最低能量构象异构体中的几何形状和振动特性的影响。计算了辛弗林的所有十二个构象异构体的振动和电子结构。辛弗林的实验 FTIR 和拉曼光谱也分别记录在光谱区域 4000-400 cm-1 和 3200-50 cm-1 中,并与辛弗林的能量最优选结构的理论光谱相关。正态坐标分析用于缩放高估的 DFT 频率并计算辛弗林振动模式的势能分布。还进行了自然键轨道分析以获得键能和占有率、HOMO-LUMO 和杂化。
更新日期:2020-01-01
中文翻译:
辛弗林及其振动和电子结构的优化
摘要 对分离的辛弗林分子及其HCl簇进行了优化。还进行了关于乙胺侧链的一些二面角的势能扫描,其优选单个辛弗林分子的十二个不同低能结构。研究了 N..H 氢键对辛弗林在最低能量构象异构体中的几何形状和振动特性的影响。计算了辛弗林的所有十二个构象异构体的振动和电子结构。辛弗林的实验 FTIR 和拉曼光谱也分别记录在光谱区域 4000-400 cm-1 和 3200-50 cm-1 中,并与辛弗林的能量最优选结构的理论光谱相关。正态坐标分析用于缩放高估的 DFT 频率并计算辛弗林振动模式的势能分布。还进行了自然键轨道分析以获得键能和占有率、HOMO-LUMO 和杂化。










































 京公网安备 11010802027423号
京公网安备 11010802027423号