当前位置:
X-MOL 学术
›
CrystEngComm
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
An insight into the non-covalent Pb⋯S and S⋯S interactions in the solid-state structure of a hemidirected lead(ii) complex†
CrystEngComm ( IF 2.6 ) Pub Date : 2019-11-11 , DOI: 10.1039/c9ce01548e Saikat Mirdya 1, 2, 3, 4, 5 , Snehasis Banerjee 5, 6, 7 , Shouvik Chattopadhyay 1, 2, 3, 4, 5
CrystEngComm ( IF 2.6 ) Pub Date : 2019-11-11 , DOI: 10.1039/c9ce01548e Saikat Mirdya 1, 2, 3, 4, 5 , Snehasis Banerjee 5, 6, 7 , Shouvik Chattopadhyay 1, 2, 3, 4, 5
Affiliation
|
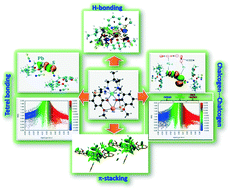
A heterodinuclear copper(II)/lead(II) complex with a compartmental ‘reduced Schiff base’ ligand (having inner N2O2 and outer  compartments) has been synthesized and characterized. Single-crystal X-ray diffraction analysis confirmed its structure. The X-ray data indicate that the inner N2O2 compartment of the compartmental reduced Schiff base is occupied by copper(II) while the outer
compartments) has been synthesized and characterized. Single-crystal X-ray diffraction analysis confirmed its structure. The X-ray data indicate that the inner N2O2 compartment of the compartmental reduced Schiff base is occupied by copper(II) while the outer  compartment is occupied by lead(II). A thiocyanate anion is coordinated to the copper(II) center via its N-end, whereas, another thiocyanate ion is linked to the lead(II) center via its S-end and is semi-coordinated to the copper(II) center through its N-end. A hemidirectionally coordinated lead(II) center is well suited for establishing tetrel bonding interactions. We estimated the BSSE (basis set superposition error) corrected energies of non-covalent S⋯S, Pb⋯π, and π⋯π interactions and N–H hydrogen bonding along with tetrel bonding by DFT calculations. To obtain an insight into the physical nature of these bonds, we extensively used Bader's quantum theory of atoms-in-molecules (QTAIM). Additionally, the non-covalent interaction reduced density gradient (NCI-RDG) method established nicely the presence of such non-covalent intermolecular interactions. Here, we also used natural bond orbital (NBO) analysis to find out the origin of S⋯S and Pb⋯S bonding.
compartment is occupied by lead(II). A thiocyanate anion is coordinated to the copper(II) center via its N-end, whereas, another thiocyanate ion is linked to the lead(II) center via its S-end and is semi-coordinated to the copper(II) center through its N-end. A hemidirectionally coordinated lead(II) center is well suited for establishing tetrel bonding interactions. We estimated the BSSE (basis set superposition error) corrected energies of non-covalent S⋯S, Pb⋯π, and π⋯π interactions and N–H hydrogen bonding along with tetrel bonding by DFT calculations. To obtain an insight into the physical nature of these bonds, we extensively used Bader's quantum theory of atoms-in-molecules (QTAIM). Additionally, the non-covalent interaction reduced density gradient (NCI-RDG) method established nicely the presence of such non-covalent intermolecular interactions. Here, we also used natural bond orbital (NBO) analysis to find out the origin of S⋯S and Pb⋯S bonding.
中文翻译:

对半定向铅(ii)配合物固态结构中非共价Pb⋯S和S⋯S相互作用的见解†
具有隔间“还原的席夫碱”配体(具有内部N 2 O 2和外部隔室)的异双核铜(II)/铅(II)配合物已被合成和表征。单晶X射线衍射分析证实了其结构。X射线数据表明,隔室还原的席夫碱的内部N 2 O 2隔室被铜(II)占据,而外部隔室被铅(II)占据。硫氰酸根阴离子通过其N端与铜(II)中心配位,而另一个硫氰酸根离子与铅(II)通过其S端居中,并通过其N端与铜(II)中心半配准。半方向协调的导线(II)中心非常适合建立Tetrel键合相互作用。我们通过DFT计算估计了非共价S⋯S,Pb⋯π和π⋯π相互作用以及N–H氢键和t铁键的BSSE(基址叠加误差)校正能量。为了深入了解这些键的物理性质,我们广泛使用了Bader的分子中原子量子理论(QTAIM)。另外,非共价相互作用降低密度梯度(NCI-RDG)方法很好地建立了这种非共价分子间相互作用的存在。在这里,我们还使用自然键轨道(NBO)分析来找出S⋯S和Pb⋯S键的起源。
更新日期:2020-01-15
compartments) has been synthesized and characterized. Single-crystal X-ray diffraction analysis confirmed its structure. The X-ray data indicate that the inner N2O2 compartment of the compartmental reduced Schiff base is occupied by copper(II) while the outer compartment is occupied by lead(II). A thiocyanate anion is coordinated to the copper(II) center via its N-end, whereas, another thiocyanate ion is linked to the lead(II) center via its S-end and is semi-coordinated to the copper(II) center through its N-end. A hemidirectionally coordinated lead(II) center is well suited for establishing tetrel bonding interactions. We estimated the BSSE (basis set superposition error) corrected energies of non-covalent S⋯S, Pb⋯π, and π⋯π interactions and N–H hydrogen bonding along with tetrel bonding by DFT calculations. To obtain an insight into the physical nature of these bonds, we extensively used Bader's quantum theory of atoms-in-molecules (QTAIM). Additionally, the non-covalent interaction reduced density gradient (NCI-RDG) method established nicely the presence of such non-covalent intermolecular interactions. Here, we also used natural bond orbital (NBO) analysis to find out the origin of S⋯S and Pb⋯S bonding.
中文翻译:
对半定向铅(ii)配合物固态结构中非共价Pb⋯S和S⋯S相互作用的见解†
具有隔间“还原的席夫碱”配体(具有内部N 2 O 2和外部隔室)的异双核铜(II)/铅(II)配合物已被合成和表征。单晶X射线衍射分析证实了其结构。X射线数据表明,隔室还原的席夫碱的内部N 2 O 2隔室被铜(II)占据,而外部隔室被铅(II)占据。硫氰酸根阴离子通过其N端与铜(II)中心配位,而另一个硫氰酸根离子与铅(
II)通过其S端居中,并通过其N端与铜(II)中心半配准。半方向协调的导线(II)中心非常适合建立Tetrel键合相互作用。我们通过DFT计算估计了非共价S⋯S,Pb⋯π和π⋯π相互作用以及N–H氢键和t铁键的BSSE(基址叠加误差)校正能量。为了深入了解这些键的物理性质,我们广泛使用了Bader的分子中原子量子理论(QTAIM)。另外,非共价相互作用降低密度梯度(NCI-RDG)方法很好地建立了这种非共价分子间相互作用的存在。在这里,我们还使用自然键轨道(NBO)分析来找出S⋯S和Pb⋯S键的起源。










































 京公网安备 11010802027423号
京公网安备 11010802027423号