当前位置:
X-MOL 学术
›
EMBO Mol. Med.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
CoA-dependent activation of mitochondrial acyl carrier protein links four neurodegenerative diseases.
EMBO Molecular Medicine ( IF 9.0 ) Pub Date : 2019-11-07 , DOI: 10.15252/emmm.201910488 Roald A Lambrechts 1 , Hein Schepers 1 , Yi Yu 1 , Marianne van der Zwaag 1 , Kaija J Autio 2 , Marcel A Vieira-Lara 3 , Barbara M Bakker 3 , Marina A Tijssen 4 , Susan J Hayflick 5 , Nicola A Grzeschik 1 , Ody Cm Sibon 1
EMBO Molecular Medicine ( IF 9.0 ) Pub Date : 2019-11-07 , DOI: 10.15252/emmm.201910488 Roald A Lambrechts 1 , Hein Schepers 1 , Yi Yu 1 , Marianne van der Zwaag 1 , Kaija J Autio 2 , Marcel A Vieira-Lara 3 , Barbara M Bakker 3 , Marina A Tijssen 4 , Susan J Hayflick 5 , Nicola A Grzeschik 1 , Ody Cm Sibon 1
Affiliation
|
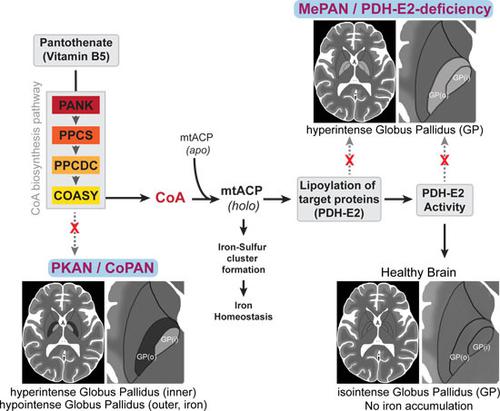
PKAN, CoPAN, MePAN, and PDH-E2 deficiency share key phenotypic features but harbor defects in distinct metabolic processes. Selective damage to the globus pallidus occurs in these genetic neurodegenerative diseases, which arise from defects in CoA biosynthesis (PKAN, CoPAN), protein lipoylation (MePAN), and pyruvate dehydrogenase activity (PDH-E2 deficiency). Overlap of their clinical features suggests a common molecular etiology, the identification of which is required to understand their pathophysiology and design treatment strategies. We provide evidence that CoA-dependent activation of mitochondrial acyl carrier protein (mtACP) is a possible process linking these diseases through its effect on PDH activity. CoA is the source for the 4'-phosphopantetheine moiety required for the posttranslational 4'-phosphopantetheinylation needed to activate specific proteins. We show that impaired CoA homeostasis leads to decreased 4'-phosphopantetheinylation of mtACP. This results in a decrease of the active form of mtACP, and in turn a decrease in lipoylation with reduced activity of lipoylated proteins, including PDH. Defects in the steps of a linked CoA-mtACP-PDH pathway cause similar phenotypic abnormalities. By chemically and genetically re-activating PDH, these phenotypes can be rescued, suggesting possible treatment strategies for these diseases.
中文翻译:

线粒体酰基载体蛋白的CoA依赖性激活将四种神经退行性疾病联系在一起。
PKAN,CoPAN,MePAN和PDH-E2缺乏症共有关键的表型特征,但在独特的代谢过程中存在缺陷。在这些遗传性神经退行性疾病中发生了对苍白球的选择性损害,这些疾病是由CoA生物合成(PKAN,CoPAN),蛋白质脂酰化(MePAN)和丙酮酸脱氢酶活性(PDH-E2缺乏)缺陷引起的。它们的临床特征重叠提示了一种常见的分子病因,需要对其进行鉴定以了解其病理生理学和设计治疗策略。我们提供的证据表明,CoA依赖的线粒体酰基载体蛋白(mtACP)激活是通过影响PDH活性将这些疾病联系起来的可能过程。CoA是翻译后4'所需的4'-磷酸鸟嘌呤部分的来源 激活特定蛋白质所需的β-磷酸泛素化。我们表明,受损的CoA稳态导致mtACP的4'-磷酸泛素化降低。这导致mtACP活性形式的减少,进而导致脂酰化的减少,而脂化蛋白(包括PDH)的活性降低。关联的CoA-mtACP-PDH途径的步骤中的缺陷会导致类似的表型异常。通过化学和遗传上的重新激活PDH,可以挽救这些表型,从而为这些疾病提出了可能的治疗策略。关联的CoA-mtACP-PDH途径的步骤中的缺陷会导致类似的表型异常。通过化学和遗传上的重新激活PDH,可以挽救这些表型,从而为这些疾病提出了可能的治疗策略。关联的CoA-mtACP-PDH途径的步骤中的缺陷会导致类似的表型异常。通过化学和遗传上的重新激活PDH,可以挽救这些表型,从而为这些疾病提出了可能的治疗策略。
更新日期:2019-12-06
中文翻译:
线粒体酰基载体蛋白的CoA依赖性激活将四种神经退行性疾病联系在一起。
PKAN,CoPAN,MePAN和PDH-E2缺乏症共有关键的表型特征,但在独特的代谢过程中存在缺陷。在这些遗传性神经退行性疾病中发生了对苍白球的选择性损害,这些疾病是由CoA生物合成(PKAN,CoPAN),蛋白质脂酰化(MePAN)和丙酮酸脱氢酶活性(PDH-E2缺乏)缺陷引起的。它们的临床特征重叠提示了一种常见的分子病因,需要对其进行鉴定以了解其病理生理学和设计治疗策略。我们提供的证据表明,CoA依赖的线粒体酰基载体蛋白(mtACP)激活是通过影响PDH活性将这些疾病联系起来的可能过程。CoA是翻译后4'所需的4'-磷酸鸟嘌呤部分的来源 激活特定蛋白质所需的β-磷酸泛素化。我们表明,受损的CoA稳态导致mtACP的4'-磷酸泛素化降低。这导致mtACP活性形式的减少,进而导致脂酰化的减少,而脂化蛋白(包括PDH)的活性降低。关联的CoA-mtACP-PDH途径的步骤中的缺陷会导致类似的表型异常。通过化学和遗传上的重新激活PDH,可以挽救这些表型,从而为这些疾病提出了可能的治疗策略。关联的CoA-mtACP-PDH途径的步骤中的缺陷会导致类似的表型异常。通过化学和遗传上的重新激活PDH,可以挽救这些表型,从而为这些疾病提出了可能的治疗策略。关联的CoA-mtACP-PDH途径的步骤中的缺陷会导致类似的表型异常。通过化学和遗传上的重新激活PDH,可以挽救这些表型,从而为这些疾病提出了可能的治疗策略。










































 京公网安备 11010802027423号
京公网安备 11010802027423号